REVIEW ARTICLE
Massoud Saidijama, Saeid Afshara, Irshad Ahmadb, Simon G. Patchingb,*
aDepartment of Molecular Medicine and Genetics, Research Centre for Molecular Medicine, School of Medicine, Hamadan University of Medical Sciences, Hamadan, Iran
bSchool of Biomedical Sciences and Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, UK
Abstract
The movement of physiologic nucleosides and nucleoside analogue drugs across biological membranes is mediated by nucleoside transport proteins. In cancer, nucleoside transporters have an important role in maintaining the hyperproliferative state of tumours and are important targets for diagnostic and therapeutic agents in the detection, treatment and monitoring of cancers. The nucleoside-based probe 3ꞌ-deoxy-3ꞌ-[18F]fluoro-L-thymidine ([18F]FLT) has been developed for PET imaging of proliferating cancer cells, which is less prone than 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) to non-specific effects. [18F]FLT enters proliferating cells through nucleoside transporters, then becomes phosphorylated and blocks DNA synthesis, whilst also becoming trapped inside the cell. Practicable and automated chemical syntheses of [18F]FLT have been developed, for which the most widely used radiolabelling precursor is the thymidine derivative 3-N-boc-5ꞌ-O-dimethoxytrityl-3ꞌ-O-nosyl-thymidine. [18F]FLT PET imaging has undergone feasibility studies and has been assessed in pre-clinical and clinical studies for the detection and diagnosis of cancers and in monitoring their response to treatments. The roles of nucleoside transporters, especially ENT1, in the cellular uptake of [18F]FLT have been investigated.
Keywords: cancer; drug delivery; [18F]FLT; hENT1; nucleoside analogues; nucleoside transport; PET imaging; radiosynthesis
1. Introduction
Nucleoside transporters as chemotherapeutic targets and biomarkers of drug response
Movement of physiologic nucleosides and hydrophilic nucleoside analogues across biological membranes is mediated by nucleoside transport proteins. Whilst physiologic nucleosides enter central salvage pathways in nucleotide biosynthesis, nucleoside analogue drugs are used in the treatment of cancer and viral diseases. In the case of cancer, nucleoside transport has an important role in maintaining the hyperproliferative state of most tumours and is therefore an important target for diagnostic and therapeutic agents in the detection, treatment and monitoring of cancers. Indeed, the clinical efficacy of anticancer nucleoside analogue drugs depends on a complex interdependence of transporters mediating entry of drugs into cells, efflux mechanisms that remove drugs from intracellular compartments and cellular metabolism to active metabolites [1–6].
In humans, two solute carrier gene families (SLC28 and SLC29) are foremost responsible for the uptake of nucleosides and nucleoside analogues into cells [7–11]. The SLC28 human concentrative nucleoside transporter (hCNT) family contains three members that mediate unidirectional transport of nucleosides into cells against their concentration gradient driven by a downward sodium gradient that moves in the same direction (symport). hCNTs are high affinity transporters found predominantly in intestinal and renal epithelia and also in other specialised cell types. hCNT1 and hCNT2 have preferences for pyrimidine and purine nucleosides, respectively, whilst hCNT3 has broad nucleoside selectivity [12–15].
The SLC29 human equilibrative nucleoside transporter (hENT) family contains four members that mediate bidirectional unenergised transport of nucleosides down their concentration gradient (facilitated diffusion). hENTs are widely distributed in most, possibly all, cell types and hENTs 1-3 have broad specificity for both purine and pyrimidine nucleosides. hENT4, also known as PMAT, is uniquely selective for adenosine and also transports a variety of organic cations [16–20]. Some nucleoside-derived drugs can also interact with and be translocated by members of the SLC22 gene family, which include organic anion transporters (OATs), organic cation transporters (OCTs) and organic carnitine and zwitterion transporters (OCTNs) [9,21–25].
The pyrimidine nucleoside analogue gemcitabine (2′,2′-difluorodeoxycytidine, trade name Gemzar) (1) is widely used as a first-line chemotherapeutic drug in the treatment of various cancers including bladder cancer, breast cancer, non-small cell lung cancer, ovarian cancer and pancreatic cancer. Unfortunately, there is often rapid development of either de novo or induced drug resistance, which significantly limits the effectiveness of gemcitabine chemotherapy.
Whilst the cytotoxic effects of gemcitabine are exerted following phosphorylation and then inhibition of DNA synthesis, it must first enter cells through nucleoside transporters, especially the ubiquitous hENT1. Hence, hENT1 expression and activity has been identified as an important prognostic biomarker in gemcitabine-treated cancers and therefore as a predictive biomarker of gemcitabine efficacy. This is particularly true of pancreatic cancer, where high expression of hENT1 is associated with increased overall survival and disease-free survival in patients treated with gemcitabine [26–30]. It therefore follows that a deficiency in hENT1 confers resistance to the cytotoxicity of gemcitabine [31–34] and approaches have been explored to overcome hENT1 deficiency. For example, upregulation of hENT1 expression in pancreatic cell lines by indole-3-carbinol enhanced the efficacy of gemcitabine [35]. Functionalised lipophilic nanoparticles have also been developed for delivery of gemcitabine into cells that bypass nucleoside transporters [36,37]. In addition to gemcitabine, other nucleoside analogues have been used and explored as chemotherapeutic drugs [38–41].
2. Pet imaging using 3ꞌ-deoxy-3ꞌ-[18F]-fluoro-L-thymidine
2.1. Overview
Positron emission tomography (PET) is a non-invasive clinical nuclear medicine technique routinely used to produce two- or three-dimensional images of the body for diagnosing and monitoring a wide range of human diseases. The PET system detects pairs of gamma rays emitted indirectly by a short-lived positron emitting radionuclide (or radiotracer), which is introduced into the body on a biologically active molecule [42]. Because PET images directly reflect in vivo tissue physiology and metabolism, one of their foremost uses is in the detection of proliferating cancer cells and monitoring their response to treatments. Indeed, the early metabolic changes associated with cancers can be detected by PET imaging before more advanced morphologic changes are detected by anatomic imaging techniques such as computed tomography (CT) and magnetic resonance imaging (MRI). This allows earlier diagnosis and earlier intervention with appropriate treatments that are more likely to have a successful outcome. By far the most commonly used radiotracer in PET imaging is 2-deoxy-2-[18F]fluoro-D-glucose ([18F]FDG) (2) [42,43]. In the case of imaging cancers, however, [18F]FDG is not necessarily the most appropriate radiotracer to use because it can accumulate non-specifically to produce false-positive findings [44]. For example, enhanced uptakes of [18F]FDG also occur in infection and in inflamed cells and lesions as well as in necrotic cells [45,46]. Alternative nucleoside-based probes that are less prone to non-specific effects have have therefore been developed for imaging tumour proliferation to use alongside [18F]FDG [44,47], the most successful being 3ꞌ-deoxy-3ꞌ-[18F]fluoro-L-thymidine ([18F]FLT) (3) [48].
2.2. Cellular trapping of [18F]FLT
Like thymidine (4), [18F]FLT is transported into cells by nucleoside transporters. Once inside the cell, [18F]FLT is a substrate for thymidine kinase I (TK1) and is phosphorylated but is not incorporated into DNA. Phosphorylated [18F]FLT cannot exit the cell and [18F]FLT is not a substrate for thymidine phosphorylase and so is not significantly degraded in vivo and is retained inside the cell (Figure 1). TK1 is a key enzyme that is upregulated in cancer cells and, in agreement with separate studies [49,50], it is assumed that the concentration of [18F]FLT inside cells is proportional to TK1 activity and therefore to cellular proliferation. One of the characteristics of tumour cells is an unchecked proliferation and it is important to measure the proliferation rate of cancer lesions to help differentiate benign from malignant tumours and to characterise malignant tumours amongst normal tissues. A further advantage of [18F]FLT is that it is only a substrate for TK1 and not for mitochondrial TK2, making it a more specific radiotracer compared with other fluorinated nucleoside analogues for cellular proliferation.
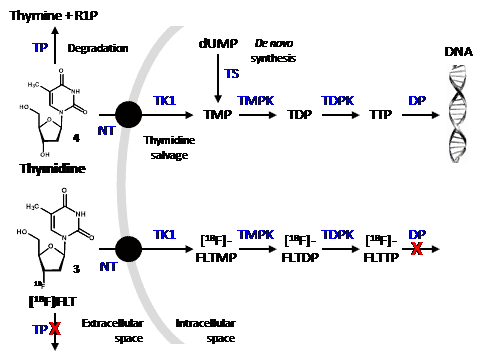
Figure 1. Cellular uptake and fate of thymidine and [18F]FLT.
In the thymidine salvage pathway, both thymidine (4) and [18F]FLT (3) undergo uptake into cells by nucleoside transporters (NT) and are initially phoshorylated by thymidine kinase 1 (TK1) and then further phosphorylated by thymidine monophosphate kinase (TMPK) and thymidine diphosphate kinase (TDPK). There is also a de novo synthesis of TMP by thymidylate synthase (TS) from deoxyuridine monophoshate (dUMP). Whilst phosphorylated thymidine is incorporated into DNA, phosphorylated [18F]FLT is not a substrate for DNA polymerase (DP) or nucleoside transporters and therefore becomes trapped inside the cell. Similarly, [18F]FLT is not a substrate for thymidine phosphorylase (TP) and so does not undergo significant degradation to thymine and ribose-1-phosphate (R1P). [18F]FLT is ultimately metabolised to its glucuronide by glucuronyl transferase in the liver and excreted by the kidney. Some cancer drugs inhibit the glucuronosyl transferase reaction, however.
2.3. Synthesis and quality control of [18F]FLT
Radiosynthesis of [18F]FLT was first reported by Wilson et al [51] using a thymidine precursor (5) with trityl and mesyl protecting groups at the 5ꞌ- and 3ꞌ-hydroxyl positions, respectively, and this was treated with [18F]potassium fluoride (Scheme 1).
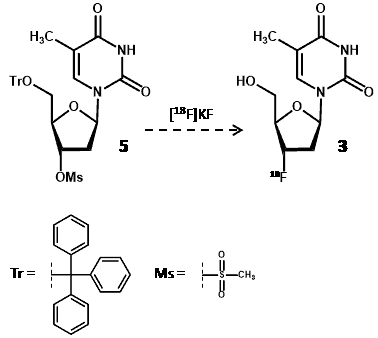
Scheme 1. Synthesis of [18F]FLT (3) using a protected thymidine radiolabelling precursor (5) and [18F]potassium fluoride.
Significant developments towards a more practicable method to produce [18F]FLT for clinical PET imaging were later made by Grierson and Shields [52–55]. Their improved method made minimal use of specialised materials and apparatus and included a three-step radiosynthesis producing [18F]FLT with a radiochemical yield (at end of bombardment) of 13% and an end of synthesis yield of 7% over 94 minutes [56]. The method used a nosylate (4-nitrobenzenesulphonate) ester as the radiolabelling precursor [1-(2-deoxy-3-O-(4-nitrobenzenesulfonyl)-5-O-(4,4ꞌ-dimethoxy-trityl)-β-D-threo-pento-furanosyl)-3-(2,4-dimethoxybenzyl)thymine] (12) that was synthesised in seven steps from thymidine (4) in an overall yield of 17% (Scheme 2).
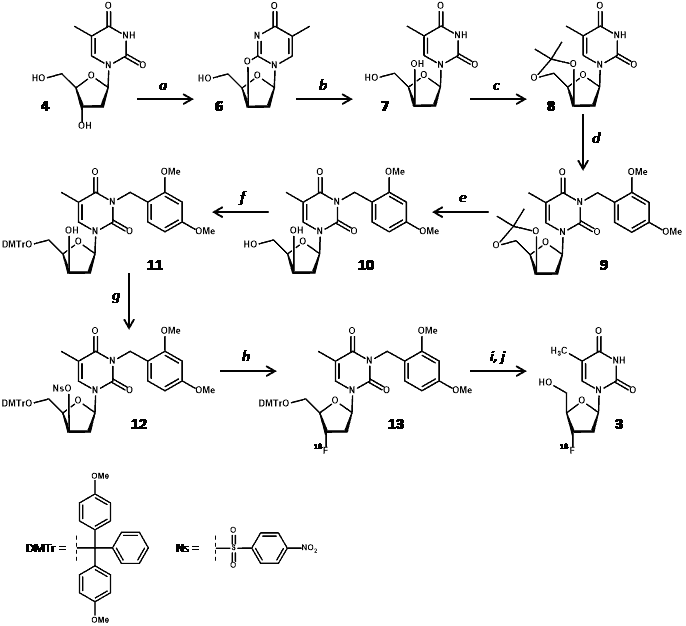
Scheme 2. Synthesis of [18F]FLT (3) from thymidine (4) using a nosylated radiolabelling precursor (12). a. 2 equiv. DIAD/TPP, MeCN, <215 °C, then H2O; b. LiOH (1 equiv.)/H2O, then H1-resin; c. acetone/PPTS (cat), reflux; d. 2,4-DMBnCl, K2CO3/MEK, reflux, phase transfer catalyst; e. EtOH-H2O, PPTS (cat), reflux; f. DMTrCl, pyr, rt; g. 4-NBS-Cl/AgOTf, pyr, 0 °C; h. K2CO3/KRY(2.2.2)/[18F]fluoride (n.c.a.), MeCN, 100 °C, 10 min; i. CAN, MeCN-EtOH-H2O (4:1:1), 100 °C, 3 min; j. C-18 HPLC.
Nucleophilic displacement of (12) with [18F]fluoride was followed by deprotection with ceramic ammonium nitrate (CAN) and then product isolation by C-18 preparative HPLC. Use of CAN for deprotection resulted in formation of precipitates such that filtration was required before HPLC, which is not conducive with synthesis automation. An alternative approach using an anhydro derivative as the radiolabelling precursor [2,3′-anhydro-5′-O-(4,4′-dimethoxytrityl)-thymidine] (14) achieved an end of synthesis [18F]FLT yield of 5.6 ± 1.4% over 90-140 minutes [57].
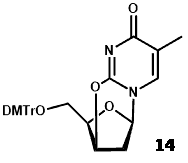
Whilst this method had a simpler precursor synthesis, radiolabelling reaction and workup of [18F]FLT, the requirement for a high boiling point reaction solvent (DMSO) compromised HPLC isolation of [18F]FLT. Using the same anhydro precursor (14) and single neutral alumina column purification, a fully automated and simplified synthesis of [18F]FLT produced an uncorrected radiochemical yield of 8.48 ± 0.93% (n = 5) in a total time of 68 ± 3 minutes and a radiochemical purity of >95% [58]. This method obviated the need for HPLC purification and the product was tested for safe levels of residual aluminium and DMSO.
New radiolabelling precursors were assessed using different protecting groups at the 5ꞌ-hydroxyl position [trityl (Tr) and 4,4′-dimethoxytrityl (DMTr)] and different electrophilic centres at the 3ꞌ-carbon [methylsulfonyl (mesyl/Ms), p-toluenesulfonyl (tosyl/Ts) and 4-nitrobenzenesulfonyl (nosyl/Ns) groups]. These precursors also had 3-N-Boc-protection, which avoided use of CAN and the resultant precipitates, thus enabling use of an automated synthesis module (Scheme 3) [59].
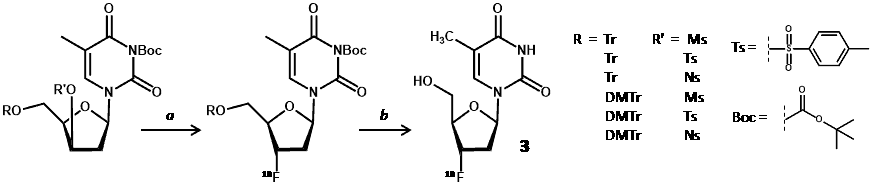
Scheme 3. Radiolabelling precursor for synthesis of [18F]FLT (3). a. [18F]fluoride, 100 °C, 10 min; b. CAN, 3 min then C-18 HPLC.
The nosylated precursors proved most successful for radiolabelling with 18F and best results were obtained using 3-N-boc-5′-O-dimethoxytrityl-3′-O-nosyl-thymidine (15) with an [18F]FLT yield (at end of bombardment) of 19.8% and an end of synthesis yield of 11.7% over 85 minutes. Using the same radiolabelling precursor (15), various [18F]fluorination and purification conditions were assessed for achieving a higher radiochemical yield of [18F]FLT [60]. Purification of the reaction mixture using an Alumina N Sep-Pak cartridge before HPLC application significantly increased the radiochemical yield to 42 ± 5.4% (decay-corrected) in under 60 min with a radiochemical purity of >97%. Again using precursor 15, a fully automated method for synthesis of [18F]FLT was developed by modifying a commercial synthesiser for [18F]FDG that uses disposable cassettes [61].
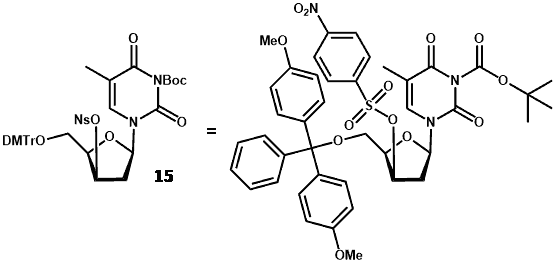
[18F]FLT yields (decay corrected) of 50.5 ± 5.2% (n = 28) and 48.7 ± 5.6% (n = 10) were obtained using 3.7 and 37.0 GBq of [18F]fluoride starting activity, respectively, in 60.0 ± 5.4 minutes including HPLC isolation. A simplified and fully automated synthesis of [18F]FLT was developed using a PET-MF-2V-IT-I [18F]FDG synthesis module by a one-pot two-step reaction procedure. The method included nucleophilic fluorination of 15 with [18F]fluoride, followed by hydrolysis of the protecting group with 1.0 M HCl in the same reaction vessel and purification with SEP PAK cartridges instead of HPLC [62]. The corrected [18F]FLT radiochemical yield was 23.2 ± 2.6% (n = 6) and the radiochemical purity was >97% obtained in a total time of 35 minutes. It was also discovered that nucleophilic fluorination of 15 using a protic solvent produced an improved radiochemical yield of [18F]FLT. Reaction in t-butanol using an automated synthesis module led to an [18F]FLT radiochemical yield of 60.2 ± 5.2% after HPLC purification [63].
The 3-N-Boc-protected compound 15 remains the most commonly used radiolabelling precursor for [18F]FLT synthesis and is commercially available at GMP grade. Indeed, [18F]FLT suitable for microPET studies has been efficiently synthesised from 15 using an electrowetting-on-dielectric digital microfluidic chip [64] and an automated and efficient radiosynthesis of [18F]FLT using a low amount of 15 (5 mg) has been developed, achieving a corrected radiochemical yield of 54% in a time of 52 minutes [65].
It is clear that the radiosynthesis of [18F]FLT can lead to many complex and potentially toxic side-products. According to the Society of Nuclear Medicine and Molecular Imaging (SNMMI), the NIH requires an [18F]FLT radiochemical purity of no less than 95% and no more than 5 mcg of nonradioactive FLT and no more than 5 mcg of other UV-absorbing impurities. [18F]FLT is therefore subject to stringent tests of quality control and biological assessment [66,67].
2.4. Feasibility studies for measuring tumour proliferation using [18F]FLT
Early prospective and feasibility studies were highly supportive of [18F]FLT as a PET radiotracer for measuring tumour proliferation, many of which performed direct comparisons of [18F]FLT and [18F]FDG uptake and correlations with immunohistochemistry results. For example, in a validation study for [18F]FLT PET imaging of proliferation in early stage non-small cell lung cancer, there was excellent correlation of [18F]FLT uptake with immunohistochemistry marker of cell proliferation Ki-67 values and flow cytometry results [68]. In a separate prospective PET study of newly diagnosed lung nodules, [18F]FLT uptake correlated significantly better with proliferation of lung tumours than did uptake of [18F]FDG, suggesting that it might be more useful as a selective biomarker for tumour proliferation [69]. PET imaging of cell proliferation in colorectal cancer using [18F]FLT and [18F]FDG showed a statistically significant positive correlation between SUVs of tumours visualised with [18F]FLT and the corresponding immunohistochemistry results, whilst no such correlation was demonstrated with [18F]FDG avid lesions [70]. In a study investigating the feasibility of [18F]FLT PET imaging for detection and grading of soft tissue sarcoma at the extremities, the method was successful in visualising cell proliferation and in differentiating between low-grade and high-grade lesions (Figure 2).
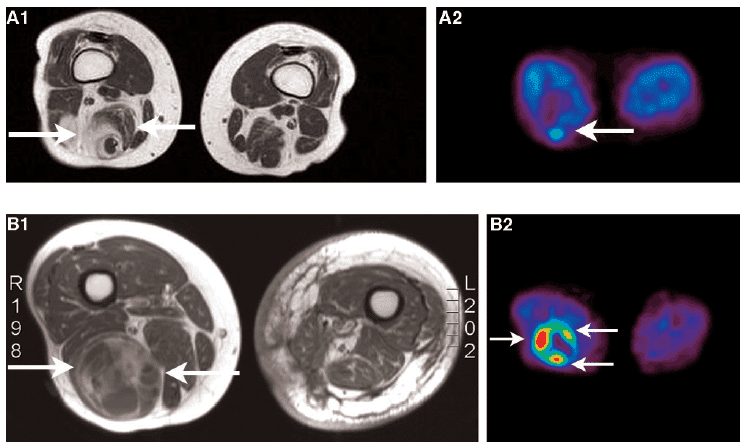
Figure 2. Images of soft tissue carcinomas. A. MRI (A1) and [18F]FLT (A2) images of a low-grade soft tissue sarcoma. B. MRI (B1) and [18F]FLT (B2) images of a high-grade soft tissue sarcoma. The MRI images of both patients demonstrate a heterogeneous tumour. [18F]FLT uptake in the high-grade soft tissue sarcoma is higher than in the low-grade soft tissue sarcoma, however. This figure was reproduced with permission from Cobben et al (2004) [71]; copyright © 2004 by American Association for Cancer Research.
The uptake of [18F]FLT correlated with the proliferation of soft tissue sarcoma [71]. In a comparative study for imaging laryngeal cancer with [18F]FLT and [18F]FDG, the numbers of cancers detected with both tracers were equal and the uptake of [18F]FDG was higher than that of [18F]FLT [72]. In a study that directly compared [18F]FLT and [18F]FDG for imaging proliferation in brain tumours of the same patients, [18F]FLT was more sensitive than [18F]FDG for imaging recurrent high-grade tumours (Figure 3), it correlated better with immunohistochemistry Ki-67 values and was a more powerful predictor of tumour progression and survival [73].

Figure 3. Images of a newly diagnosed glioblastoma. A. MRI image (contrast-enhanced T1-weighted image) showing a large area of contrast enhancement in the right frontal lobe. Both [18F]FDG PET (B) and [18F]FLT PET (C) show increased uptake in same area. This figure was reproduced with permission from Chen et al (2005) [73]; copyright © 2005 by Society of Nuclear Medicine and Molecular Imaging.
More recently, a study investigating the performance of cellular metabolism PET imaging with [18F]FDG versus cellular proliferation imaging with [18F]FLT for detecting cervical lymph node metastases in oral/head and neck cancer was performed. Whilst [18F]FLT showed better overall performance for detecting lymphadenopathy on qualitative assessment within the total nodal population, [18F]FDG performed better for pathologic discrimination within the visible lymph nodes [74]. [18F]FLT PET imaging has been assessed in a range of further pre-clinical and clinical studies for the detection and diagnosis of cancers and in monitoring their response to treatments. A comprehensive overview of these studies is beyond the scope of this work, so the reader is referred to recent review articles on this theme [75–80].
3. Nucleoside transporters in [18F]FLT Pet imaging of cancer
A number of studies have investigated the roles of nucleoside transporters, especially hENT1, in measuring cell proliferation of cancers using [18F]FLT PET. One of the first studies implicating a role for ENT1 in uptake of [18F]FLT into cancer cells was an assessment of [18F]FLT PET imaging for early measurement of thymidylate synthase inhibition in tumours [81]. Radiation-induced fibrosarcoma-1 tumor-bearing mice, injected with the thymidylate synthase inhibitor 5-fluorouracil, were imaged by [18F]FLT PET 1 to 2 hours after treatment (Figure 4).
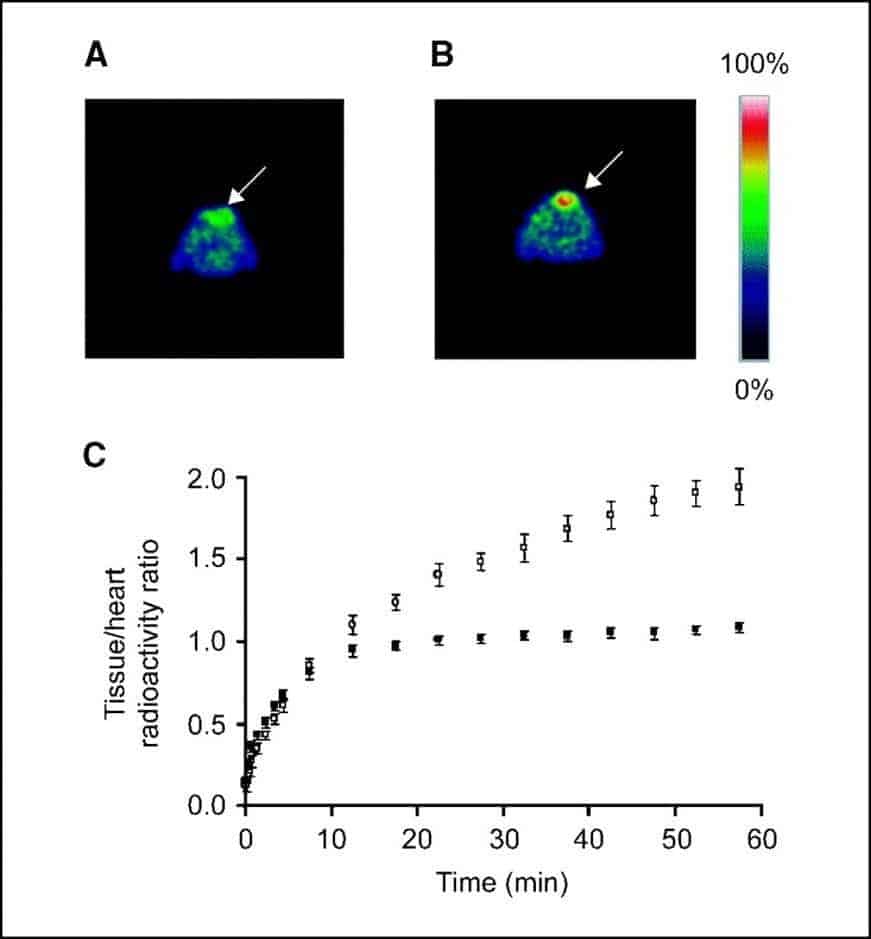
Figure 4. [18F]FLT PET imaging of thymidylate synthase inhibition in tumours. Typical 0.5-mm transverse [18F]FLT PET slices through the thoracic region at the level of the maximum tumour diameter of a RIF-1 tumour-bearing mouse treated with PBS (control; A) and a RIF-1 tumour-bearing mouse treated with 5-fluorouracil (B). Arrows = tumour. C. Summary of [18F]FLT kinetics in control (•) and 5-fluorouracil-treated (○) RIF-1 tumours. Tumour-bearing mice were treated with PBS or 5-fluorouracil at a dose of 165 mg/kg i.p. and scanned at 1 to 2 hours after injection. For each mouse, tumour/heart radioactivity ratios from five slices were averaged at each of the 19 time points. Data points represent mean tumour/heart ratios from eight control mice and five 5-fluorouracil-treated mice; error bars represent standard errors. This figure was reproduced with permission from Perumal et al (2006) [81]; copyright © 2006 by American Association for Cancer Research.
Parallel measurements included whole-cell assays implicating a functional role for ENT1, in which there was an increase in ENT1-binding sites per cell from 49,110 in untreated cells to 73,142 (P = 0.03) in cells treated with 5-fluorouracil (10 mg/ml, 2 hours), without a change in transporter affinity (P = 0.41). It was concluded that [18F]FLT PET can measure thymidylate synthase inhibition as early as 1 to 2 hours after treatment with 5-fluorouracil by a mechanism involving redistribution of ENT1to the plasma membrane [81].
In a study specifically designed to investigate roles of human nucleoside transporters in uptake of FLT [82], binding of FLT to transporters was initially monitored by its inhibitory effects on [3H]uridine (1 mM) uptake in yeast cells producing recombinant transporters. The lowest FLT Ki value for inhibition of [3H]uridine uptake was produced by hCNT1, followed by hCNT3, hENT2, hENT1 and hCNT2. Transport of [3H]FLT (20 mM) into Xenopus laevis oocytes individually producing recombinant nucleoside transporters produced uptake values of 48 ± 8, 32 ± 5, 12 ± 1, 11 ± 0.8 and 2.0 ± 0.2 pmol/oocyte/30 min for hCNT1, hCNT3, hENT2, hENT1 and hCNT2, respectively (Figure 5A). Transport of [3H]FLT by hENT1, hENT2, hCNT1 and hCNT3 was concentration-dependent and conformed to Michaelis-Menten kinetics (Figure 5B). hENT1 and hENT2 produced higher transport capacities and lower apparent affinities than hCNT1 and hCNT3. The transport efficiency (Vmax/Km) was approximately 6-fold greater for hCNT1 and hCNT3 than for hENT1 and hENT2, suggesting that hCNT1 and hCNT3 transport [3H]FLT more efficiently than hENT1 and hENT2 at lower (micromolar) concentrations [82]. [3H]FLT uptake in six different cancer cell lines was inhibited at least 50% by the hENT1 inhibitor nitrobenzylmercaptopurine ribonucleoside (NBMPR) and, according to real-time polymerase chain reactions, hENT1 and hENT2 had the most abundant nucleoside transporter transcripts in all cell lines. Further binding assays demonstrated a strong correlation between extracellular NBMPR binding sites/cell and [3H]FLT uptake for all but one of the cell lines, consistent with plasma membrane nucleoside transporters (especially hENT1) having important roles in cellular FLT uptake [82].
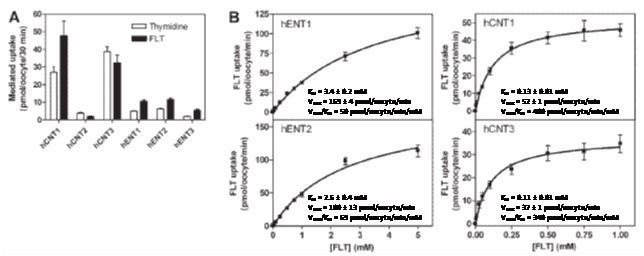
Figure 5. Uptake of FLT by human nucleoside transporters. A. Uptake of [3H]thymidine and [3H]FLT (20 mM) in Xenopus laevis oocytes producing different recombinant human nucleoside transport proteins. B. Concentration-dependent influx of [3H]FLT in oocytes producing human nucleoside transport proteins with inset kinetic parameters. All experiments were performed with 12 oocytes per group and data are expressed as mean S.E.M. Error bars are not shown if the S.E.M. values were smaller than the size of the symbol. Values are for mediated uptake (uptake in RNA transcript-injected oocytes minus uptake in control oocytes injected with water alone). Pictures were reproduced with permission from Paproski et al (2008) [82]; copyright © 2008 by American Society for Pharmacology and Experimental Therapeutics.
A subsequent study investigated the importance of ENT1 for [18F]FLT uptake in normal tissues and tumours [83]. ENT1-knockout (ENT1(-/-)) mice were compared with wild-type (ENT1(+/+)) mice using small-animal [18F]FLT PET in absence and presence of NBMPR-phosphate (Figure 6). Compared with noninjected ENT1(+/+) mice, ENT1(+/+) mice injected with NBMPR-P and ENT1(-/-) mice displayed a reduced percentage injected dose per gram (%ID/g) for [18F]FLT in the blood (84% and 81%, respectively) and an increased %ID/g for [18F]FLT in the spleen (188% and 469%, respectively) and bone marrow (266% and 453%, respectively).
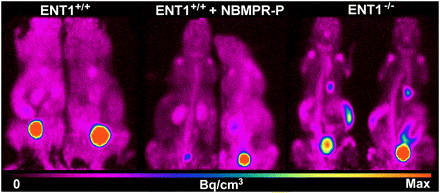
Figure 6. Role of ENT1 in cellular uptake of [18F]FLT. [18F]FLT PET maximum-intensity-projection images of noninjected ENT1+/+ mice, ENT1+/+ mice injected with 15 mg of NBMPR-P per kg at 1 hour before imaging, and ENT1−/− mice. Images were summations of radioactivity over 10 minutes from approximately 50 to 60 minutes after radiotracer injection. This figure was reproduced with permission from Paproski et al (2010) [83]; copyright © 2010 by Society of Nuclear Medicine.
Plasma thymidine levels were 1.65-fold higher in ENT1(-/-) mice than in ENT1(+/+) mice, whilst spleen tissue from ENT1(+/+) and ENT1(-/-) mice showed similar TK1 protein levels and significant staining of CNT1 and CNT3 [83]. Human lung carcinoma cells transfected with pSUPER-producing short-hairpin RNA against hENT1 (A549-pSUPER-hENT1) displayed 0.45-fold hENT1 transcript levels and 0.68-fold [3H]FLT uptake compared with cells transfected with a scrambled sequence with no homology to mammalian genes (A549-pSUPER-SC). Compared with A549-pSUPER-SC xenograft tumors, A549-pSUPER-hENT1 xenograft tumors displayed 0.76-fold %ID/g values (ex vivo gamma-counts) and 0.65-fold maximum SUV (PET image analysis) for [18F]FLT uptake at 1 h after tracer injection. Because loss of ENT1 activity significantly affected [18F]FLT biodistribution in mice and [18F]FLT uptake in xenograft tumors, it was concluded that ENT1 is an important mediator of [18F]FLT uptake in normal tissues and tumours [83].
A parallel study was performed to determine if FLT uptake is a predictor of gemcitabine uptake and/or toxicity in a panel of six different human pancreatic cancer cell lines (Capan-2, AsPC-1, BxPC-3, PL45, MIA PaCa-2 and PANC-1) [84]. Capan-2 cells displayed the lowest levels of extracellular NBMPR binding, FLT and gemcitabine uptake during short (1-45 seconds) and prolonged (1 hour) periods, and gemcitabine sensitivity. Exposure to NBMPR (inhibits only hENT1) or dilazep (inhibits hENT1 and hENT2) reduced FLT and gemcitabine uptake and gemcitabine sensitivity, with dilazep having greater effects than NBMPR. Gemcitabine permeation was primarily mediated by hENT1, and to a lesser extent by hENT2, whilst FLT permeation included a substantial component of passive diffusion [84]. In five out of six cell lines, correlations were observed between FLT and gemcitabine initial rates of uptake, gemcitabine uptake and gemcitabine toxicity, FLT uptake and gemcitabine toxicity, and ribonucleotide reductase subunit M1 expression and gemcitabine toxicity. Uptakes of FLT and gemcitabine were comparable for predicting gemcitabine toxicity in the tested pancreatic cancer cell lines, it was therefore concluded that [18F]FLT may provide clinically useful information about tumour gemcitabine transport capacity and sensitivity [84].
In a study investigating the correlation of [18F]FLT uptake with mRNA expressions of hENT1 and TK1 in tissue samples from newly diagnosed gastrointestinal cancers, of all lesions tested only one gastric cancer showed focally increased uptake of [18F]FLT. The mean [18F]FLT SUV in gastrointestinal cancer was 5.48 ± 1.87. No significant correlation was observed between [18F]FLT SUV and hENT1 mRNA expression (P = 0.90), whilst there was a significant correlation between [18F]FLT SUV and TK1 mRNA expression (P <0.05) [85].
Isolated human B-lymphobast cells, either proficient or deficient in TK1, were studied to show how metabolism and nucleoside transport influence uptake and retention of FLT [86]. Both influx and efflux of FLT were measured under conditions where concentrative and equilibrative transport could be distinguished. Whilst initial rates of FLT uptake were a function of both concentrative and equilibrative transporters, concentrative FLT transport dominated over equilibrative transport. Inhibition of hENT1 reduced FLT uptake, but there were no correlations between clonal variations in hENT1 levels and FLT uptake. TK1 was mandatory for the cellular concentration of FLT and uptake peaked after 60 minutes of incubation with FLT, followed by a decline in intracellular levels of FLT and its metbolites. Efflux was rapid and was associated with reductions in FLT and its metabolites [86].
In a study examining the extent to which ENT1 levels vary in a proliferation-dependent manner in human A549 tumor cells grown as tumor xenografts in nude mice, [18F]FLT uptake was measured in vivo using small animal PET and further examined ex vivo using autoradiography [87]. [18F]FLT uptake patterns were also compared to immunohistochemical analysis of ENT1 and the proliferation markers Ki67 and BrdU. ENT1 levels were approximately twice as high in actively proliferating regions of tumours grown in vivo. Proliferating regions showed increased [18F]FLT uptake compared with nonproliferating tumour regions, hence confirming the role of hENT1 in [18F]FLT uptake and strengthening the case for using [18F]FLT as a tracer for both cell proliferation and relative ENT1 levels [87].
A later study investigated whether uptake of [18F]FLT in newly diagnosed gliomas correlates with ENT1 mRNA expression, microvascular density (assessed by CD34 immunohistochemistry) and blood-brain barrier permeability [88]. In tumour lesions identified by increased [18F]FLT uptake, dynamic analysis revealed correlations between the phosphorylation rate constant k3 and ENT1 expression, but there was no correlation between the kinetic parameters and CD34 score. Good correlation was observed between the gadolinium (Gd) enhancement score (evaluating blood-brain barrier breakdown) and ENT1 expression, CD34 score and Ki-67 index. It was therefore concluded that ENT1 expression might not reflect accumulation of [18F]FLT in vivo due to blood-brain barrier permeability in glioma [88].
TAS-102 is a recently developed orally administered combination chemotherapy drug composed of α,α,α-trifluorothymidine (TFT) and a thymidine phosphorylase inhibitor (tipiracil hydrochloride, TPI) in a 1:0.5 ratio. TAS-102 has especially been targeted at metastatic colorectal cancer [89–94]. In the mechanism of action of TAS-102, TFT is intracellularly phosphorylated and then incorporated into DNA, which leads to DNA damage and cell cycle arrest. TPI is an inhibitor of thymidine phosphorylase that metabolises TFT, therefore increasing the bioavailability of TFT, and TPI is also an inhibitor of angiogenesis. hCNT1 has a major role in intestinal absorption of TFT and, when expressed in Xenopus laevis oocytes, uptake of TFT by hCNT1 has Km and Vmax values of 69.0 μM and 516 pmol/oocyte/30 min, respectively [95]. In human colon cancer xenografts in mice, administration of TAS-102 imparted a decrease in cell viability and an increase in [18F]FLT uptake. Early after TAS-102 administration there may be decreased dephosphorylation of [18F]FLT and, at a later time, increased TK1 expression and/or nucleoside transporter activity may be related to increased [18F]FLT uptake. Hence, [18F]FLT PET is potentially useful for assessing the pharmacodynamics of TAS-102 in cancer patients [96].
4. Conclusions
The nucleoside analogue [18F]FLT is emerging as a feasible radiotracer for routine PET imaging, especially in the detection and monitoring of cancers. The important advantage of [18F]FLT is that it suffers from a lower non-specific background uptake than the established and widely used radiotracer [18F]FDG. Practicable and automated chemical syntheses of [18F]FLT have been developed, for which the most widely used radiolabelling precursor is the thymidine derivative 3-N-boc-5ꞌ-O-dimethoxytrityl-3ꞌ-O-nosyl-thymidine. [18F]FLT enters proliferating cells through nucleoside transporters, which are also routes of entry into cells for anti-cancer and anti-viral nucleoside analogue drugs. The roles of nucleoside transporters, especially ENT1, in the cellular uptake of [18F]FLT have been investigated. Further studies on structure-activity relationships and regulation of nucleoside transporters are necessary for improving the design and delivery of nucleoside analogue drugs and for ongoing developments in PET imaging of cancers and other diseases.
Conflict of interest
The authors confirm that this article content has no conflict of interest.
Funding
This work was supported by the Hamadan University of Medical Sciences and the University of Leeds.
References
Key References: 2, 9, 30, 47, 48, 56, 68, 77, 78, 81
- Damaraju VL, Damaraju S, Young JD, Baldwin SA, Mackey J, Sawyer MB, Cass CE. Nucleoside anticancer drugs: the role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene. 2003;22(47): 7524-7536.CrossRef PubMed
- King AE, Ackley MA, Cass CE, Young JD, Baldwin SA. Nucleoside transporters: from scavengers to novel therapeutic targets. Trends Pharmacol Sci. 2006;27(8): 416-425.CrossRef PubMed
- Zhang J, Visser F, King KM, Baldwin SA, Young JD, Cass CE. The role of nucleoside transporters in cancer chemotherapy with nucleoside drugs. Cancer Metastasis Rev. 2007;26(1): 85-110.CrossRef PubMed
- Molina-Arcas M, Casado FJ, Pastor-Anglada M. Nucleoside transporter proteins. Curr Vasc Pharmacol. 2009;7(4): 426-434. CrossRef PubMed
- Hagmann W, Jesnowski R, Löhr JM. Interdependence of gemcitabine treatment, transporter expression, and resistance in human pancreatic carcinoma cells. Neoplasia. 2010;12(9): 740-747.CrossRef PubMed
- Choi JS, Berdis AJ. Nucleoside transporters: biological insights and therapeutic applications. Future Med Chem. 2012;4(11): 1461-1478. CrossRef PubMed
- Molina-Arcas M, Trigueros-Motos L, Casado FJ, Pastor-Anglada M. Physiological and pharmacological roles of nucleoside transporter proteins. Nucleosides Nucleotides Nucleic Acids. 2008;27(6): 769-778.CrossRef PubMed
- Young JD, Yao SY, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med. 2013;34(2-3): 529-547.CrossRef PubMed
- Pastor-Anglada M, Pérez-Torras S. Nucleoside transporter proteins as biomarkers of drug responsiveness and drug targets. Front Pharmacol. 2015;6: 13. CrossRef
- Dos Santos-Rodrigues A, Grañé-Boladeras N, Bicket A, Coe IR. Nucleoside transporters in the purinome. Neurochem Int. 2014;73: 229-237.CrossRef PubMed
- Young JD. The SLC28 (CNT) and SLC29 (ENT) nucleoside transporter families: a 30-year collaborative odyssey. Biochem Soc Trans. 2016;44(3): 869-876. CrossRef PubMed
- Gray JH, Owen RP, Giacomini KM. The concentrative nucleoside transporter family, SLC28. Pflugers Arch. 2004;447(5): 728-734.CrossRef PubMed
- Aymerich I, Duflot S, Fernández-Veledo S, Guillén-Gómez E, Huber-Ruano I, Casado FJ, Pastor-Anglada M. The concentrative nucleoside transporter family (SLC28): new roles beyond salvage? Biochem Soc Trans. 2005;33(Pt 1): 216-219.CrossRef PubMed
- Pastor-Anglada M, Cano-Soldado P, Errasti-Murugarren E, Casado FJ. SLC28 genes and concentrative nucleoside transporter (CNT) proteins. Xenobiotica. 2008;38(7-8): 972-994.CrossRef PubMed
- Sun L, Xie H, Ingram JC, Hope RJ, Young JD, Baldwin SA, Patching SG. Characterization of the Escherichia coli concentrative nucleoside transporter NupC using computational, biochemical and biophysical methods. Biochemistry. 2017 [Epub ahead of print].CrossRef PubMed
- Baldwin SA, Beal PR, Yao SY, King AE, Cass CE, Young JD. The equilibrative nucleoside transporter family, SLC29. Pflugers Arch. 2004;447(5): 735-743.CrossRef PubMed
- Zhou M, Xia L, Engel K, Wang J. Molecular determinants of substrate selectivity of a novel organic cation transporter (PMAT) in the SLC29 family. J Biol Chem. 2007;282(5): 3188-3195.CrossRef PubMed
- Young JD, Yao SY, Sun L, Cass CE, Baldwin SA. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica. 2008;38(7-8): 995-1021.CrossRef PubMed
- Boswell-Casteel RC, Hays FA. Equilibrative nucleoside transporters-A review. Nucleosides Nucleotides Nucleic Acids. 2017;36(1): 7-30.CrossRef PubMed
- Saidijam M, Karimi Dermani F, Sohrabi S, Patching SG. Efflux proteins at the blood-brain barrier: review and bioinformatics analysis. Xenobiotica. 2017 [Epub ahead of print]CrossRef PubMed
- Cano-Soldado P, Pastor-Anglada M. Transporters that translocate nucleosides and structural similar drugs: structural requirements for substrate recognition. Med Res Rev. 2012;32(2): 428-457.CrossRef PubMed
- Arimany-Nardi C, Errasti-Murugarren E, Minuesa G, Martinez-Picado J, Gorboulev V, Koepsell H, Pastor-Anglada M. Nucleoside transporters and human organic cation transporter 1 determine the cellular handling of DNA-methyltransferase inhibitors. Br J Pharmacol. 2014;171(16): 3868-3880.CrossRef PubMed
- Zhu C, Nigam KB, Date RC, et al. Evolutionary analysis and classification of OATs, OCTs, OCTNs, and other SLC22 transporters: Structure-function implications and analysis of sequence motifs. PLoS One. 2015;10(11): e0140569.CrossRef PubMed
- Drenberg C, Gibson AA, Pounds S, et al. OCTN1 is a high-affinity carrier of nucleoside analogs. Cancer Res. 2017;77(8): 2102-2111.CrossRef PubMed
- Gesto DS, Cerqueira NM, Fernandes PA, Ramos MJ. Gemcitabine: a critical nucleoside for cancer therapy. Curr Med Chem. 2012;19(7): 1076-1087.CrossRef PubMed
- Nakano Y, Tanno S, Koizumi K, et al. Gemcitabine chemoresistance and molecular markers associated with gemcitabine transport and metabolism in human pancreatic cancer cells. Br J Cancer. 2007;96(3): 457-463.CrossRef PubMed
- Farrell JJ, Elsaleh H, Garcia M, et al. Human equilibrative nucleoside transporter 1 levels predict response to gemcitabine in patients with pancreatic cancer. Gastroenterology. 2009;136(1): 187-195.CrossRef PubMed
- Spratlin JL, Mackey JR. Human equilibrative nucleoside transporter 1 (hENT1) in pancreatic adenocarcinoma: Towards individualized treatment decisions. Cancers (Basel). 2010;2(4): 2044-2054.CrossRef PubMed
- Mohelnikova-Duchonova B, Melichar B. Human equilibrative nucleoside transporter 1 (hENT1): do we really have a new predictive biomarker of chemotherapy outcome in pancreatic cancer patients? Pancreatology. 2013;13(6): 558-563.CrossRef PubMed
- Nordh S, Ansari D, Andersson R. hENT1 expression is predictive of gemcitabine outcome in pancreatic cancer: a systematic review. World J Gastroenterol. 2014;20(26): 8482-8490.
- Mackey JR, Jennings LL, Clarke ML, et al. Immunohistochemical variation of human equilibrative nucleoside transporter 1 protein in primary breast cancers. Clin Cancer Res. 2002;8(1): 110-116.PubMed
- Reiman T, Clarke ML, Dabbagh L, et al. Differential expression of human equilibrative nucleoside transporter 1 (hENT1) protein in the Reed-Sternberg cells of Hodgkin’s disease. Leuk Lymphoma. 2002;43(7): 1435-1440.CrossRef PubMed
- Andersson R, Aho U, Nilsson BI, Peters GJ, Pastor-Anglada M, Rasch W, Sandvold ML. Gemcitabine chemoresistance in pancreatic cancer: molecular mechanisms and potential solutions. Scand J Gastroenterol. 2009;44(7): 782-786.CrossRef PubMed
- Chow L, Lai R, Dabbagh L, Belch A, Young JD, Cass CE, Mackey JR. Analysis of human equilibrative nucleoside transporter 1 (hENT1) protein in non-Hodgkin’s lymphoma by immunohistochemistry. Mod Pathol. 2005;18(4): 558-564.CrossRef PubMed
- Wang H, Word BR, Lyn-Cook BD. Enhanced efficacy of gemcitabine by indole-3-carbinol in pancreatic cell lines: the role of human equilibrative nucleoside transporter 1. Anticancer Res. 2011;31(10): 3171-3180.PubMed
- Ansari D, Chen BC, Dong L, Zhou MT, Andersson R. Pancreatic cancer: translational research aspects and clinical implications. World J Gastroenterol. 2012;18(13): 1417-1424. PubMed
- Lansakara-P DS, Rodriguez BL, Cui Z. Synthesis and in vitro evaluation of novel lipophilic monophosphorylated gemcitabine derivatives and their nanoparticles. Int J Pharm. 2012;429(1-2): 123-134. Erratum in Int J Pharm. 2012;438(1-2): 336.CrossRef
- Galmarini CM, Mackey JR, Dumontet C. Nucleoside analogues and nucleobases in cancer treatment. Lancet Oncol. 2002;3(7): 415-424.CrossRef PubMed
- Galmarini CM, Jordheim L, Dumontet C. Pyrimidine nucleoside analogs in cancer treatment. Expert Rev Anticancer Ther. 2003;3(5): 717-728.CrossRef PubMed
- Szafraniec SI, Stachnik KJ, Skierski JS. New nucleoside analogs in the treatment of solid tumors. Acta Pol Pharm. 2004;61(4): 297-305.PubMed
- Jordheim LP, Durantel D, Zoulim F, Dumontet C. Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat Rev Drug Discov. 2013;12(6): 447-464.CrossRef PubMed
- Patching SG (2015) Roles of facilitative glucose transporter GLUT1 in [18F]FDG positron emission tomography (PET) imaging of human diseases. J. Diagn. Imaging Ther. 2015;2(1): 30-102.CrossRef
- Fanti S, Farsad M, Mansi L. Normal distribution of FDG In: Atlas of PET/CT – A quick guide to image interpretation. Berlin Heidelberg: Springer; 2009: 1-23. Reference Source
- Alauddin MM. Nucleoside-based probes for imaging tumor proliferation using positron emission tomography. J Labelled Comp Radiopharm. 2013;56(3-4): 237-243.CrossRef PubMed
- Kubota R, Yamada S, Kubota K, Ishiwata K, Tamahashi N, Ido T. Intratumoral distribution of fluorine-18-fluorodeoxyglucose in vivo: High accumulation in macrophages and granulation tissues studied by microautoradiography. J Nucl Med. 1992;33(11): 1972-1980.PubMed
- van Waarde A, Cobben DC, Suurmeijer AJ, et al. Selectivity of 18F-FLT and 18F-FDG for differentiating tumor from inflammation in a rodent model. J Nucl Med. 2004;45(4): 695-700.PubMed
- Alauddin MM, Gelovani JG. Pyrimidine nucleosides in molecular PET imaging of tumor proliferation. Curr Med Chem. 2010;17(11): 1010-1029.CrossRef PubMed
- Shields AF, Grierson JR, Dohmen BM, et al. Imaging proliferation in vivo with [F-18]FLT and positron emission tomography. Nat Med. 1998;4(11): 1334-1336.CrossRef PubMed
- Sherley JL, Kelly TJ. Thymidine kinase I (TK1) activity is thought to be proportional to cellular proliferation and DNA synthesis by the salvage pathway. J Biol Chem. 1988;263(17): 8350-8358.PubMed
- Al-Madhoun AS, Tjarks W, Eriksson S. The role of thymidine kinases in the activation of pyrimidine nucleoside analogues. Mini Rev Med Chem. 2004;4(4): 341-350.CrossRef PubMed
- Wilson IK, Chatterjee S, Wolf W. Synthesis of 3′-fluoro-3′-deoxythymidine and studies of its 18F-radiolabeling, as a tracer for the noninvasive monitoring of the biodistribution of drugs against AIDS. J Fluorine Chem. 1991;55(3): 283-289.CrossRef
- Grierson JR, Shields AF, Eary JF. Development of a radiosynthesis for 3′-[18F]fluoro-3′-deoxy-nucleosides. J Labelled Compd Radiopharm. 1997;40(1): 60-62.Reference Source
- Grierson JR, Shields AF. Developments in the radiosynthesis of [18F]FLT. J Nucl Med. 1998;39(5): 22P.PubMed
- Grierson JR, Shields AF. An improved synthesis of [18F]FLT. J Labelled Compd Radiopharm. 1999;42(suppl 1): S525-S526.
- Grierson JR, Shields AF. Optimizing labeling substrate structure for 3′-deoxy-3′-[18F]fluorothymidine: [18F]FLT. J Nucl Med. 1999;40(5): 83P.
- Grierson JR, Shields AF. Radiosynthesis of 3′-deoxy-3′-[(18)F]fluorothymidine: [(18)F]FLT for imaging of cellular proliferation in vivo. Nucl Med Biol. 2000;27(2): 143-156.CrossRef PubMed
- Wodarski C, Eisenbarth J, Weber K, Henze M, Haberkorn U, Eisenhut M. Synthesis of 3′-deoxy-3′-[18F]fluoro-thymidine with 2,3-anhydro-5′-O-(4,4′-dimethoxytrityl)-thymidine. J Labelled Compd Radiopharm. 2000;43(12): 1211-1218.
- Nandy SK, Rajan MJR. Fully automated and simplified radiosynthesis of [18F]-3′-deoxy-3′-fluorothymidine using anhydro precursor and single neutral alumina column purification. J Radioanal Nucl Chem. 2010;283(3): 741-748.CrossRef
- Martin SJ, Eisenbarth JA, Wagner-Utermann U, et al. A new precursor for the radiosynthesis of [18F]FLT. Nucl Med Biol. 2002;29(2) :263-273.CrossRef PubMed
- Yun M, Oh SJ, Ha HJ, Ryu JS, Moon DH. High radiochemical yield synthesis of 3′-deoxy-3′-[18F]fluorothymidine using (5′-O-dimethoxytrityl-2′-deoxy-3′-O-nosyl-beta-D-threo pentofuranosyl)thymine and its 3-N-BOC-protected analogue as a labeling precursor. Nucl Med Biol. 2003;30(2): 151-157.CrossRef PubMed
- Oh SJ, Mosdzianowski C, Chi DY, et al. Fully automated synthesis system of 3′-deoxy-3′-[18F]fluorothymidine. Nucl Med Biol. 2004;31(6): 803-809.CrossRef PubMed
- Tang G, Tang X, Wen F, Wang M, Li B. A facile and rapid automated synthesis of 3′-deoxy-3′-[18F]fluorothymidine. Appl Radiat Isot. 2010;68(9): 1734-1739.CrossRef PubMed
- Lee SJ, Oh SJ, Chi DY, Kil HS, Kim EN, Ryu JS, Moon DH. Simple and highly efficient synthesis of 3′-deoxy-3′-[18F]fluorothymidine using nucleophilic fluorination catalyzed by protic solvent. Eur J Nucl Med Mol Imaging. 2007;34(9): 1406-1409.CrossRef PubMed
- Javed MR, Chen S, Kim H-K, et al. Efficient radiosynthesis of 3′-deoxy-3′-[18F]fluorothymidine using electrowetting-on-dielectric digital microfluidic chip. J Nucl Med. 2014;55(2): 321-328.CrossRef PubMed
- Marchand P, Ouadi A, Pellicioli M, Schuler J, Laquerriere P, Boisson F, Brasse D. Automated and efficient radiosynthesis of [18F]FLT using a low amount of precursor. Nucl Med Biol. 2016;43: 520-527.CrossRef PubMed
- Jiang X, Wang XX, Lu H, et al. Quality control and biological assessment of automated synthesized 3′-deoxy-3′-18F-fluorothymidine using CFN-MPS-200 Module. Sichuan Da Xue Xue Bao Yi Xue Ban. 2017;48(2): 295-298.PubMed
- Nascimento LTC, Silveira MB, Ferreira SMZMD, Silva JB. Comparison between two ethanolic solutions for 3′-deoxy-3′-[18F]fluorothymidine elution. Adv Chem Eng Sci. 2017;7: 23-33.CrossRef
- Vesselle H, Grierson J, Muzi M, et al. In vivo validation of 3’deoxy-3′-[(18)F]fluorothymidine ([(18)F]FLT) as a proliferation imaging tracer in humans: correlation of [(18)F]FLT uptake by positron emission tomography with Ki-67 immunohistochemistry and flow cytometry in human lung tumors. Clin Cancer Res. 2002;8(11): 3315-3323.PubMed
- Buck AK, Halter G, Schirrmeister H, et al. Imaging proliferation in lung tumors with PET: 18F-FLT versus 18F-FDG. J Nucl Med. 2003;44(9): 1426-1431.PubMed
- Francis DL, Freeman A, Visvikis D, et al. In vivo imaging of cellular proliferation in colorectal cancer using positron emission tomography. Gut. 2003;52(11): 1602-1606.CrossRef PubMed
- Cobben DC, Elsinga PH, Suurmeijer AJ, Vaalburg W, Maas B, Jager PL, Hoekstra HJ. Detection and grading of soft tissue sarcomas of the extremities with (18)F-3′-fluoro-3′-deoxy-L-thymidine. Clin Cancer Res. 2004;10(5): 1685-1690.CrossRef PubMed
- Cobben DC, van der Laan BF, Maas B, et al. 18F-FLT PET for visualization of laryngeal cancer: comparison with 18F-FDG PET. J Nucl Med. 2004;45(2): 226-231.PubMed
- Chen W, Cloughesy T, Kamdar N, et al. Imaging proliferation in brain tumors with 18F-FLT PET: comparison with 18F-FDG. J Nucl Med. 2005;46(6): 945-952.PubMed
- Schaefferkoetter JD, Carlson ER, Heidel RE. Can 3′-deoxy-3′-((18)F) fluorothymidine out perform 2-deoxy-2-((18)F) fluoro-D-glucose positron emission tomography/computed tomography in the diagnosis of cervical lymphadenopathy in patients with oral/head and neck cancer? J Oral Maxillofac Surg. 2015;73(7): 1420-1428.CrossRef PubMed
- Salskov A, Tammisetti VS, Grierson J, Vesselle H. FLT: measuring tumor cell proliferation in vivo with positron emission tomography and 3′-deoxy-3′-[18F]fluorothymidine. Semin Nucl Med. 2007;37(6): 429-439. Erratum in Semin Nucl Med. 2008;38(2): 148.CrossRef
- Jensen MM, Kjaer A. Monitoring of anti-cancer treatment with (18)F-FDG and (18)F-FLT PET: a comprehensive review of pre-clinical studies. Am J Nucl Med Mol Imaging. 2015;5(5): 431-456.PubMed
- Peck M, Pollack HA, Friesen A, et al. Applications of PET imaging with the proliferation marker [18F]-FLT. Q J Nucl Med Mol Imaging. 2015;59(1): 95-104.PubMed
- Bollineni VR, Kramer GM, Jansma EP, Liu Y, Oyen WJ. A systematic review on [(18)F]FLT-PET uptake as a measure of treatment response in cancer patients. Eur J Cancer. 2016;55: 81-97.CrossRef PubMed
- Schelhaas S, Heinzmann K, Bollineni VR, et al. Preclinical applications of 3′-deoxy-3′-[18F]fluorothymidine in oncology – A systematic review. Theranostics. 2017;7(1): 40-50.CrossRef PubMed
- Nikaki A, Angelidis G, Efthimiadou R, et al P. 18F-fluorothymidine PET imaging in gliomas: an update. Ann Nucl Med. 2017;31(7): 495-505.CrossRef PubMed
- Perumal M, Pillai RG, Barthel H, et al. Redistribution of nucleoside transporters to the cell membrane provides a novel approach for imaging thymidylate synthase inhibition by positron emission tomography. Cancer Res. 2006;66(17): 8558-8564.CrossRef PubMed
- Paproski RJ, Ng AM, Yao SY, Graham K, Young JD, Cass CE. The role of human nucleoside transporters in uptake of 3′-deoxy-3′-fluorothymidine. Mol Pharmacol. 2008;74(5): 1372-1380.CrossRef PubMed
- Paproski RJ, Wuest M, Jans HS et al. Biodistribution and uptake of 3′-deoxy-3′-fluorothymidine in ENT1-knockout mice and in an ENT1-knockdown tumor model. J Nucl Med. 2010;51(9): 1447-1455.PubMed
- Paproski RJ, Young JD, Cass CE. Predicting gemcitabine transport and toxicity in human pancreatic cancer cell lines with the positron emission tomography tracer 3′-deoxy-3′-fluorothymidine. Biochem Pharmacol. 2010;79(4): 587-595.CrossRef PubMed
- Kameyama R, Yamamoto Y, Izuishi K, Sano T, Nishiyama Y. Correlation of 18F-FLT uptake with equilibrative nucleoside transporter-1 and thymidine kinase-1 expressions in gastrointestinal cancer. Nucl Med Commun. 2011;32(6): 460-465.CrossRef PubMed
- Plotnik DA, McLaughlin LJ, Chan J, Redmayne-Titley JN, Schwartz JL. The role of nucleoside/nucleotide transport and metabolism in the uptake and retention of 3′-fluoro-3′-deoxythymidine in human B-lymphoblast cells. Nucl Med Biol. 2011;38(7): 979-986.CrossRef PubMed
- Plotnik DA, Asher C, Chu SK, et al. Levels of human equilibrative nucleoside transporter-1 are higher in proliferating regions of A549 tumor cells grown as tumor xenografts in vivo. Nucl Med Biol. 2012;39(8): 1161-1166.CrossRef PubMed
- Shinomiya A, Miyake K, Okada M, et al. 3′-Deoxy-3′-[(18)F]-fluorothymidine ([(18)F]-FLT) transport in newly diagnosed glioma: correlation with nucleoside transporter expression, vascularization, and blood-brain barrier permeability. Brain Tumor Pathol. 2013;30(4): 215-223.CrossRef PubMed
- Lenz HJ, Stintzing S, Loupakis 3. TAS-102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev. 2015;41(9): 777-783.CrossRef PubMed
- Miyamoto Y, Lenz HJ, Baba H. A novel antimetabolite: TAS-102 for metastatic colorectal cancer. Expert Rev Clin Pharmacol. 2016;9(3): 355-365.CrossRef PubMed
- Mota JM, Fonseca LG, Braghiroli MI, Hoff PM. Review on TAS-102 development and its use for metastatic colorectal cancer. Crit Rev Oncol Hematol. 2016;104: 91-97.CrossRef PubMed
- Uboha N, Hochster HS. TAS-102: a novel antimetabolite for the 21st century. Future Oncol. 2016;12(2): 153-163.CrossRef PubMed
- Marcus L, Lemery SJ, Khasar S, et al. FDA Approval Summary: TAS-102. Clin Cancer Res. 2017;23(12): 2924-2927.CrossRef PubMed
- Puthiamadathil JM, Weinberg BA. Emerging combination therapies for metastatic colorectal cancer – impact of trifluridine/tipiracil. Cancer Manag Res. 2017;9: 461-469.CrossRef PubMed
- Takahashi K, Yoshisue K, Chiba M, Nakanishi T, Tamai I. Involvement of concentrative nucleoside transporter 1 in intestinal absorption of trifluridine using human small intestinal epithelial cells. J Pharm Sci. 2015;104(9): 3146-3153.CrossRef PubMed
- Lee HJ, Oh SJ, Lee EJ, Chung JH, Kim Y, Ryu JS, Kim SY, Lee SJ, Moon DH, Kim TW. Positron emission tomography imaging of human colon cancer xenografts in mice with [18F]fluorothymidine after TAS-102 treatment. Cancer Chemother Pharmacol. 2015;75(5): 1005-1013.CrossRef PubMed
Article information
Corresponding author: Simon G. Patching.
Copyright: © 2018 Saidijam M, Afshar F, Ahmad I, Patching SG.
How to cite: Saidijam M, Afshar F, Ahmad I, Patching SG. Nucleoside transporters in PET imaging of proliferating cancer cells using 3ꞌ-deoxy-3ꞌ-[18F]fluoro-L-thymidine. J. Diagn. Imaging Ther. 2018; 5(1):1-13 (https://dx.doi.org/10.17229/jdit.2018-0210-030).
Article history: Received 15 December 2017; Accepted 01 February 2018; Published online 10 February 2018.
Archive link: 2018-0210-030
home »