REVIEW ARTICLE
Simon G. Patching
School of Biomedical Sciences and the Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
Abstract
This review concerns methods of synthesis, NMR analysis and applications of isotope labelled hydantoins. The hydantoin moiety is present in natural products and in extraterrestrial ice, indicating this to be an important compound in prebiotic chemistry. Bacterial transport proteins that scavenge hydantoins have been identified, isolated and characterised with isotope-labelling of hydantoins as an essential requirement to achieve this. These are Mhp1 from Microbacterium liquefaciens and PucI from Bacillus subtilis, transporting 5-aryl-substituted hydantoins and allantoin, respectively. The hydantoin ring is a useful centre in synthetic chemistry, especially for combinatorial chemistry, multicomponent reactions and in diversity-oriented synthesis. It is also found in pharmacologically active molecules, such as the anticonvulsant phenytoin. Hydantoins synthesised with isotope labels include hydantoin itself, allantoin, other 5-monosubstituted derivatives, phenytoin, other 5,5-di-substituted derivatives, N-substituted derivatives and other more complex molecules with multiple substituents. Analysis of isotope-containing hydantoins by NMR spectroscopy has been important for confirming purity, labelling integrity, specific activity and molecule conformation. Isotope labelled hydantoins have been used in a range of biological, biomedical, food and environmental applications including metabolic and in vivo tissue distribution studies, biochemical analysis of transport proteins, identification and tissue distribution of drug binding sites, drug metabolism and pharmacokinetic studies and as an imaging agent.
Keywords: allantoin; drug binding and metabolism; hydantoins; isotopic labeling; NMR analysis; PET imaging; phenytoin; transport assays
1. Introduction
Hydantoin (IUPAC name imidazolidine-2,4-dione) (1) (Figure 1) is a heterocyclic ring system that occurs relatively rarely in nature. The most commonly known natural product with the hydantoin ring is the urea derivative allantoin (5-ureidohydantoin) (2) (Figure 1), which is a constituent of urine and a major metabolic intermediate in most types of organisms including bacteria, fungi, plants and animals. Allantoin is also present in a number of toothpastes, mouth washes, shampoos and cosmetic products and is used in medications that treat skin conditions including acne, impetigo, eczema and psoriasis. Other natural products that contain the hydantoin ring as part of their chemical structure have been isolated from marine sponges [1,2], from a Mediterranean Sea anemone [3] and from the fungus Fusarium sp [4]. A fulvic acid polymer isolated from a coastal pond in Antarctica is also suggested to contain a hydantoin ring based on a solid-state NMR 15N and 13C{14N} chemical shift investigation [5]. Interestingly, the presence of hydantoin in extraterrestrial ice has been demonstrated, indicating this to be an important compound in prebiotic chemistry [6], and a new route for the prebiotic synthesis of hydantoin in water/ice/urea solutions involving the photochemistry of acetylene has been proposed [7].
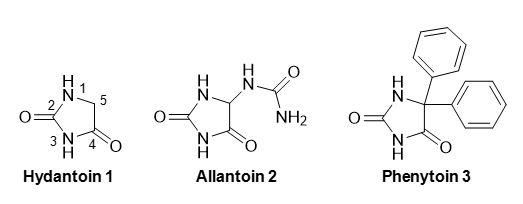
Figure 1. Structures of hydantoin (1) and the common 5-substituted derivatives allantoin (2) and phenytoin (3).
The first reported synthesis of hydantoin was in 1861 by Baeyer [8], but its structure was not assigned correctly until 1870 by Strecker [9]. The chemical properties, methods of synthesis and reactivity of hydantoin and its derivativies have been reviewed extensively [10–15]. For synthetic chemistry applications, hydantoin is a useful centre in combinatorial chemistry [16], multicomponent reactions [17,18] and in diversity-oriented synthesis [19–21]. Hydantoins substituted at the 5-position are precursors to optically pure natural and unnatural α-amino acids, which is achieved through their chemical or enzymatic hydrolysis [22–30]. They therefore serve as important compounds in the food industry, for example in the production of the artificial sweetener aspartame (N-(L-α-aspartyl)-L-phenylalanine, 1-methyl ester), which can be synthesised from its constituent L-α-amino acids. They are important in the pharmaceutical industry as precursors to optically pure D-amino acids [31–33], which are used in the production of certain drugs such as β-lactam antibiotics (e.g. penicillin and amoxicillin) and anticancer agents (e.g. goserelin). The hydantoin moiety itself also forms the basis or is a constituent of a number of pharmacologically active molecules, the most well known being anticonvulsants such as phenytoin (5,5-diphenylhydantoin) (3) (Figure 1) [34–37].
A protein called Mhp1 that promotes the uptake of 5-aryl substituted hydantoins into cells of the Gram positive bacterium Microbacterium liquefaciens, serving as part of a salvage pathway for carbon nutrients, has been identified, isolated and purified and its high-resolution crystal structure (Figure 2A) determined in three different conformations (open to outside, occluded with substrate, open to inside), the first for any secondary active transport protein [38–43]. Mhp1 is a member of the widespread nucleobase-cation-symport-1 (NCS1) family of secondary active transport proteins with members in bacteria, fungi and plants [44–52] and has provided a pivotal model for the alternating access mechanism of membrane transport and for the mechanism of ion-coupling [41,42,53–57]. Mhp1 is located in the cytoplasmic cell membrane where it catalyses the inward co-transport of a sodium ion down its concentration gradient and of a hydantoin molecule against its concentration gradient (Figure 2A). This mechanism enables the bacterium to scavenge low concentrations of hydantoin compounds from its environment. The principal transported substrates of Mhp1 are L-5-benzylhydantoin (4) and L-5-indolylmethylhydantoin (5) (Figure 2B).
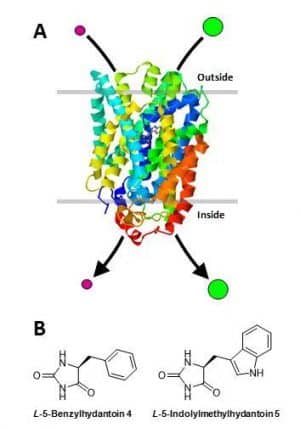
Figure 2. Hydantoin transport protein, Mhp1, from the Gram positive bacterium Microbacterium liquefaciens.
A. Schematic illustration of the 3.4 Å-resolution crystal structure of Mhp1 determined in complex with the substrate L-5-indolylmethylhydantoin [62]. Mhp1 is shown in a cell membrane where it catalyses the inward co-transport of a sodium ion (purple circle) down its concentration gradient and of a hydantoin molecule (green circle) against its concentration gradient. This mechanism enables the bacterium to scavenge low concentrations of hydantoin compounds from its environment for use as sources of carbon and nitrogen. The locations of a sodium ion and of a hydantoin molecule can be seen in the centre of the structure at their respective binding sites. The structure of Mhp1 was drawn using RSCB Protein Data Bank file 4D1A using Jmol [63]. B. Substrates of the Mhp1 transport protein L-5-benzylhydantoin (4) and L-5-indolylmethylhydantoin (5).
An allantoin transport protein called PucI from Bacillus subtilis has recently been isolated, purified and characterised [58]. PucI shares evolutionary relationships with other putative bacterial allantoin permeases, with Mhp1 and with other characterised NCS1 transporters in fungi and plants [58]. Crucial to the success in characterising the ligand recognition, substrate selectivity and transport kinetics of Mhp1 and PucI was synthesis of a number of isotope-labelled hydantoins and their use in biochemical assays [39,58–62]. Analysis of the synthesised isotope-containing hydantoin compounds by NMR spectroscopy was important for confirming their purity and labelling integrity. A significant number and variety of other isotope-labelled hydantoins have been synthesised and used in a range of biological, biomedical, food and environmental applications. The methods of synthesis, NMR analysis and their applications are the subject of this review.
2. Hydantoin
A classic method for the synthesis of hydantoin uses glycine as the starting compound (Figure 3, Scheme 1). Glycine (6) is reacted with ethanol/acid to give the ester (7); the amine group is then reacted with potassium cyanate to give the intermediate (8), which is cyclised under acid reflux to give hydantoin (1). Starting with [1-13C], [2-14C] or [15N]glycine (6a, 6b, 6c) this method has been used to prepare [4-13C], [5-14C] and [1-15N]hydantoin (1a, 1b, 1c), respectively [64–66]. The [4-13C]hydantoin and [1-15N]hydantoin were reacted with indole-3-aldehyde followed by hydrolysis to give DL-tryptophan labelled with 13C at the 1-position [64] or with 15N at the α-N position [66], respectively. The [5-14C]hydantoin was reacted with furfural (9) to give [5-14C]5-(2-furylidene)hydantoin (10) and reduced to [5-14C]5-(2-furyl)hydantoin (11), which was hydrolysed to DL-[2-14C]3-(2′-furyl)-alanine (Figure 3, Scheme 1) [65]. Another classic method for the synthesis of hydantoin, and derivatives thereof substituted at the 5-position, is the Bucherer-Bergs reaction [67–69]. This is the multicomponent reaction of carbonyl compounds (aldehydes or ketones) or cyanohydrins with potassium cyanide and ammonium carbonate to give hydantoins where the chemical groups on the carbonyl compound become the substituents at the 5-position in the hydantoin (Figure 3, Scheme 2). Use of the simplest aldehyde formaldehyde in this reaction does give hydantoin (1), but also other products including hydantoic acid and hydantoic amide [70]. Work by Winstead et al [71] nicely demonstrates the range of aliphatic, aromatic and cyclic substituted hydantoins that can be produced by the Bucherer-Bergs reaction, which in this case were labelled with the positron emitting carbon-11 at the 4-position by using [11C]potassium cyanide in the reaction (Figure 3, Scheme 3). The 11C-labelled hydantoins prepared here were used to measure their in vivo tissue distribution pattern in dogs.
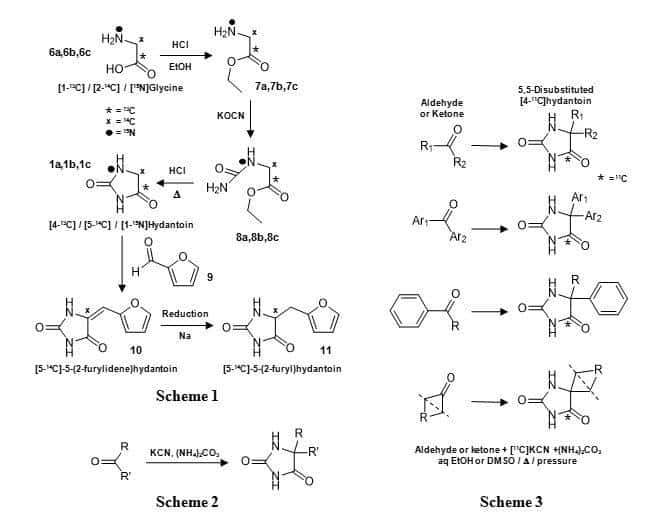
Figure 3. Synthesis of [4-13C], [5-14C] and [1-15N]hydantoin (1a, 1b, 1c) from [1-13C], [2-14C] or [15N]glycine (Scheme 1), the Bucherer-Bergs reaction for synthesis of 5-substituted hydantoins (Scheme 2) and its use in preparing a range of aliphatic, aromatic and cyclic substituted [4-11C]hydantoins (Scheme 3).
3. 5-Monosubstituted hydantoins
A number of 5-monosubstituted hydantoins have been synthesised with isotope labels, where the label has been directed to the substituent group and/or to the hydantoin moiety. Starting with [1-14C]phenol (12a), 5-(4-methoxy)benzylhydantoin (16) and 5-(4-hydroxy)benzyl-hydantoin (17) were synthesised with a 14C label at the 4-position in the benzyl ring (Figure 4, Scheme 4) [72].
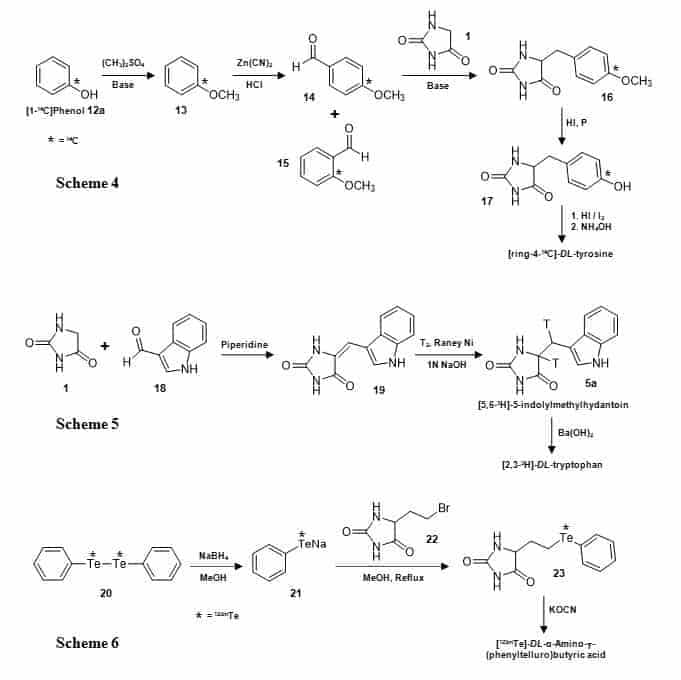
Figure 4. Synthesis of the 5-substituted hydantoins [ring-4-14C]-5-(4-methoxy)benzylhydantoin (16) and [ring-4-14C]-5-(4-hydroxy)benzylhydantoin (17) (Scheme 4), [5,6-3H]-5-indolylmethylhydantoin (5a) (Scheme 5) and [123mTe]-5-[β-(phenyltelluro)ethyl]hydantoin (23) (Scheme 6).
The [1-14C]phenol (12a) was methylated with dimethyl sulphate to give [1-14C]anisole (13) which was converted to a mixture of ortho– and para-[4-14C]-anisaldehyde (14, 15) by a modified Gattermann reaction and then the separated para-form was condensed with hydantoin (1) to give [ring-4-14C]-5-(4-methoxy)benzyl-hydantoin (16) from which the methyl group was removed to give [ring-4-14C]-5-(4-hydroxy)benzylhydantoin (17). The latter compound was then converted into [ring-4-14C]-DL-tyrosine. Hydantoins 16 and 17 have also been prepared with a 14C label at the 6-position (CH2) by condensing hydantoin with [1-14C]anisaldehyde and used as intermediates in the synthesis of [3-14C]-DL-tyrosine [73,74]. A similar approach has been used for synthesis of [1-14C]-DL-tyrosine [75,76]. A tritiated form of 5-indolylmethylhydantoin (5a) has been prepared by combining hydantoin (1) with indole-3-carboxaldehyde (18) and the resultant alkene (19) then reduced with tritium gas to give [5,6-3H]-5-indolylmethylhydantoin (5a) (Figure 4, Scheme 5). This was then converted into [2,3-3H]-DL-tryptophan [77]. Using a method based on Gaudry’s synthesis [78], another 14C-labelled amino acid, [1-14C]-L-lysine, was prepared via the intermediates 5-hydroxyhydantoin and 5-bromobutylhydantoin [79]. A hydantoin with a 123m-tellurium labelled 5-substituent (23) has been synthesised starting from [123mTe]diphenyl ditelluride (20) (Figure 4, Scheme 6). This compound was reduced by sodium borohydride to generate [123mTe]phenyltellurol (21), which was then reacted with 5-(β-bromoethyl)hydantoin (22) to give [123mTe]-5-[β-(phenyltelluro)ethyl]hydantoin (23). The hydantoin was hydrolysed to [123mTe]-DL-α-amino-g-(phenyltelluro)-butyric acid, which was used as a potential pancreatic imaging agent [80].
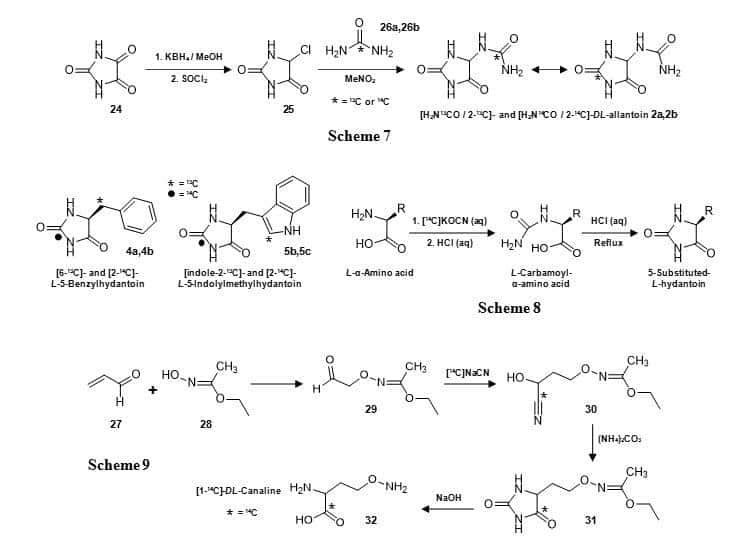
Figure 5. Synthesis of the 5-substituted hydantoins DL-[H2N13CO/13C-2]allantoin (2a) and DL-[H2N14CO/14C-2]allantoin (2b) (Scheme 7), [6-13C]-L-5-benzylhydantoin (4a), [2-14C]-L-5-benzylhydantoin (4b), [indole-2-13C]-L-5-indolylmethylhydantoin (5b), [2-14C]-L-5-indolylmethylhydantoin (5c) (Scheme 8) and a hydantoin derivative of DL-canaline with a 14C label at C-4 (31) (Scheme 9).
DL-Allantoin (2) has been synthesised with 13C or 14C labels using urea as the source of the label (Figure 5, Scheme 7) [59]. This synthesis began by reduction of parabanic acid (24) to give 5-hydroxyhydantoin, which was then treated with thionyl chloride to give 5-chlorohydantoin (25). Reaction with [13C]- or [14C]urea (26a, 26b) produced the labelled forms of DL-allantoin (2a, 2b). In this case, NMR analysis was not only important for demonstrating high purity of the labelled product, but the 13C NMR spectrum of 2a (Figure 6A) also revealed a partial scrambling of the 13C label from the ureido group (157.7 ppm) to the C-2 position (157.1 ppm) [59]. This confirmed a rearrangement of allantoin in solution via a putative bicyclic intermediate [81] and so 2a was assigned as DL-[H2N13CO/13C-2]allantoin. The 14C-labelled allantoin (2b) synthesised by the same method was therefore also assigned as DL-[H2N14CO/14C-2]allantoin [59]. The 14C-labelled compound has been used in whole cell uptake assays with the transport proteins Mhp1 and PucI in experiments to define their substrate selectivities, ligand recognition and transport kinetics (Figure 7) [58,62].
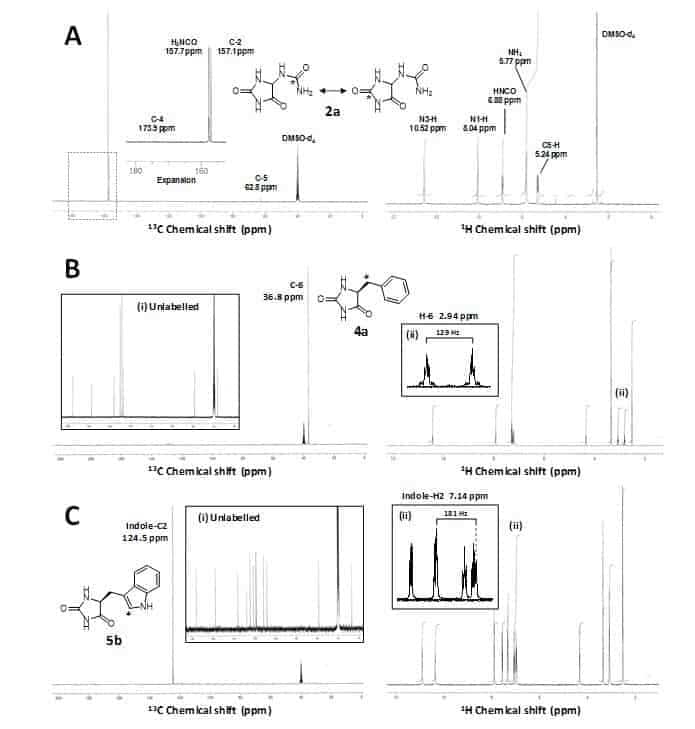
Figure 6. NMR analysis of 13C-labelled 5-substituted hydantoins.
A. 13C (left) and 1H (right) NMR spectra of DL-[H2N13CO/13C-2]allantoin (2a) in DMSO-d6 obtained using a 300 MHz magnet; inset is an expansion of a region of the 1H NMR spectrum (dotted line) containing the 13C-enriched carbonyl signals H2N13CO and 13C-2. B. 13C (left) and 1H (right) NMR spectra of [6-13C]-L-5-benzylhydantoin (4a) in DMSO-d6 obtained using a 300 MHz and 500 MHz magnet, respectively; inset are a 13C NMR spectrum of unlabelled L-5-benzylhydantoin (i) and an expansion of a region of the 1H NMR spectrum containing signals for the H-6 methylene position (ii). C. 13C (left) and 1H (right) NMR spectra of [indole-2-13C]-L-5-indolylmethylhydantoin (5b) in DMSO-d6 obtained using a 300 MHz and 500 MHz magnet, respectively; inset are a 13C NMR spectrum of unlabelled L-5-indolylmethylhydantoin (i) and an expansion of a region of the 1H NMR spectrum containing signals for the indole-H2 position (ii). This figure was constructed using the results of Patching [59,60]; copyright © 2009, 2010 by John Wiley & Sons, Ltd.
Crucially, these measurements provided the first experimental evidence to demonstrate that PucI is a medium-affinity transporter of allantoin. The Mhp1 substrates L-5-benzylhydantoin (4) and L-5-indolylmethylhydantoin (5) have been synthesised containing both 13C and 14C labels (Figure 5) for use in solid-state NMR measurements of ligand binding (unpublished) and in whole cell transport assays, respectively, with the Mhp1 protein [60,62]. These compounds were prepared from the appropriate L-α-amino acid (phenylalanine or tryptophan) by reaction with potassium cyanate under acidic conditions to give the L-carbamoyl-L-α-amino acid, which was then cyclised to give the 5-substituted L-hydantoin (Figure 5, Scheme 8). This is based on the classic Urech hydantoin synthesis [82]. [6-13C]-L-5-Benzylhydantoin (4a) was prepared from [3-13C]-L-phenylalanine and [indole-2-13C]-L-5-indolylmethyl-hydantoin (5b) was prepared from [indole-2-13C]-L-tryptophan. NMR analysis of the 13C-labelled compounds was important for confirming both high purity and labelling integrity [60]. The 13C NMR spectrum of [6-13C]-L-5-benzylhydantoin (4a) confirmed 13C enrichment exclusively in the C-6 position at 36.8 ppm and the 1H NMR spectrum showed a splitting of the H-6 methylene signal at 2.94 ppm with a coupling constant of 129 Hz due to the directly attached 13C label (Figure 6B). Similarly, the 13C NMR spectrum of [indole-2-13C]-L-5-indolylmethylhydantoin (5b) confirmed 13C enrichment exclusively in the indole-C2 position at 124.5 ppm and the 1H NMR spectrum showed a splitting of the indole-H2 signal at 7.14 ppm with a coupling constant of 181 Hz due to the directly attached 13C label (Figure 6C). The 14C-labelled versions of the compounds (4b, 5c) were prepared by using [14C]potassium cyanate in the reaction to introduce the label at C-2 in the hydantoin ring [60]. These 14C-compounds have been used in whole cell uptake assays with Mhp1 that have been crucial in defining its substrate selectivity, ligand recognition and quantitation of ligand binding and in screening the transport activities of Mhp1 mutants (Figure 7). This work also identified a novel inhibitor of Mhp1, 5-(2-naphthylmethyl)hydantoin, which was itself synthesised with a 14C label at the C-2 position using the same method (Figure 7) [62].
A hydantoin derivative of DL-canaline has been synthesised with a 14C label at C-4 in the hydantoin ring (31) as an intermediate in the production of [14C]-DL-canaline (32) itself, which is a structural analog of ornithine (Figure 5, Scheme 9) [83]. The synthesis began by reacting acrolein (27) and ethyl N-hydroxyacetimidate (28) to give ethyl N-[3-oxopropoxy]acetimidate (29), which is converted into the nitrile (30) using [14C]sodium cyanide. The nitrile was cyclised with ammonium carbonate to give the 14C-labelled hydantoin (31). Heating of the hydantoin with sodium hydroxide afforded [1-14C]-DL-canaline (32), which was intended for use in evaluating its capacity to support amino acid biosynthesis by a seed-eating beetle.
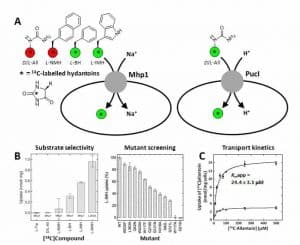
Figure 7. Whole-cell uptake assays for the bacterial transport proteins Mhp1 and PucI using 14C-labelled hydantoins.
A. Schematic illustration of an assay measuring the uptake of 14C-labelled hydantoins into energised Escherichia coli cells with amplified expression of the transport proteins Mhp1 from Microbacterium liquefaciens (left) and PucI from Bacillus subtilis (right). The 14C-labelled hydantoins tested with sodium-dependent Mhp1 and with proton-dependent PucI are D/L-allantoin (D/L-All, 2b), L-5-(2-naphthylmethyl)hydantoin (L-NMH), L-5-benzylhydantoin (L-BH, 4b) and L-5-indolylmethylhydantoin (L-IMH, 5c). The colours represent transported compounds (green) and non-transported compounds (red). B. Results for characterising the substrate selectivity of Mhp1 using radiolabelled L-tryptophan (L-Trp) and the 14C-labelled hydantoins listed above (left) and for screening mutants of Mhp1 for transport of [14C]-L-IMH (5c) compared with wild-type (right). C. Results for the concentration-dependence of initial rate [14C]-D/L-allantoin (2b) uptake into cells that were uninduced (open circles) or induced (closed circles) for expression of PucI. The data were fitted to the Michaelis-Menten equation to derive the given rate constant (Kmapp) for PucI-mediated transport. Pictures in B were reproduced from Simmons et al (2014) [62]; copyright © 2014 by the authors. Results in C were reproduced from Ma et al (2016) [58]; copyright © 2016 by the authors.
4. Phenytoin and derivatives
The hydantoin that has been isotope-labelled and used in biomedical applications with highest proliferation is the anticonvulsant drug phenytoin (5,5-diphenylhydantoin) (3). A wide range of different isotope labels, labelling patterns and synthetic routes have been used with phenytoin and with derivatives of phenytoin.
Phenytoin with all three carbon positions in the hydantoin ring 13C-labeled has been synthesised with [13C]carbon dioxide and [13C]urea as the sources of the labels (Figure 8, Scheme 10) [84]. Reaction of the Grignard reagent phenylmagnesium bromide (33) with [13C]carbon dioxide to give [13CO]benzoic acid (34) was followed by thionyl chloride treatment to give [13CO]benzoyl chloride (35a) and then reduction to give [13CO]benzaldehyde (36a). Coupling of two benzaldehyde molecules by reaction with sodium cyanide gave [13CO,13C-OH]benzoin (37a), which was reacted with [13C]urea (26a) to give [2,4,5-13C]-5,5-diphenylhydantoin (3a). In this case the compound was synthesised for use as a stable-isotope labelled biomedical tracer using mass spectrometry for detection; this was prior to the availability of a radiolabelled form of phenytoin. Later, both Emran et al [85] and Iida et al [86] used the same reaction of benzoin (37b) with 11C- or 13C-labelled urea (26a, 26b) to produce [2-11C]- or [2-13C]-5,5-diphenylhydantoin (3b, 3c), respectively (Figure 8, Scheme 11).
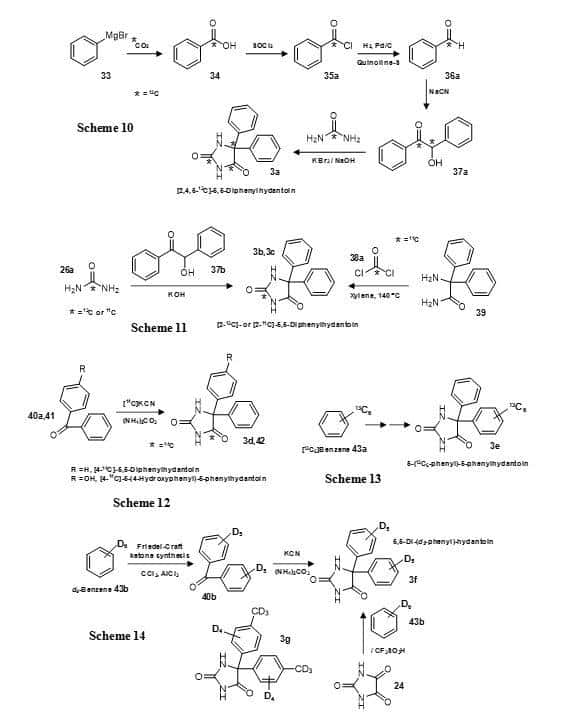
Figure 8. Synthesis of isotope-labelled versions of phenytoin (5,5-diphenylhydantoin) (3) (Schemes 10-14).
[2-11C]-5,5-Diphenylhydantoin (3c) has also been prepared by reacting 2-amino-2,2-diphenylacetamide (39) with [11C]phosgene (38a) (Figure 8, Scheme 11) [87]. The Bucherer-Bergs reaction has been used with benzophenone (40a) and [14C]potassium cyanide to produce [4-14C]-5,5-diphenylhydantoin (3d) and the same method also used to prepare its major metabolite [4-14C]-5-(4-hydroxyphenyl)-5-phenylhydantoin (42) (Figure 8, Scheme 12) [88]. A modification of the same reaction with [11C]hydrogen cyanide was also used to produce 5,5-diphenylhydantoin and 5-(4-hydroxyphenyl)-5-phenylhydantoin with an 11C label at the 4-position and these were used in measurements of their in vivo distribution [89]. The brain and whole-body pharmacokinetics of the 11C-labelled 5,5-diphenylhydantoin in rats has been evaluated using a planar positron imaging system (Figure 9), which demonstrated a difference in the brain distribution of the compound between intravenous and duodenal administration [90].
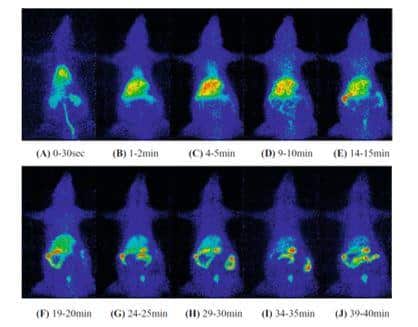
Figure 9. Whole-body imaging of 11C-labelled 5,5-diphenylhydantoin (DPH) injected into tail vein of rat using planar positron imaging.
A tracer amount of 11C-DPH (1.5 μg, approximately 5 MBq/250 g) was injected into the tail vein and scans were performed over a total period of 40 minutes. Summation images were created for the first 30 seconds (A) and for the given 1 minute intervals (B–J) to show the accumulation and changes in distribution of 11C-DPH over time. This figure was reproduced with permission from Hasegawa et al (2008) [90]; copyright © 2008 by The Japanese Society of Nuclear Medicine.
[13C6]Benzene (43a) has been subject to Friedel-Crafts reaction and then followed by Bucherer-Bergs reaction to produce 5-(13C6-phenyl)-5-phenylhydantoin (3e) (Figure 8, Scheme 13) [91]. Using a similar approach but starting with d6-benzene (43b), a deuterated form of phenytoin has been synthesised with all aromatic hydrogen atoms replaced by deuterium (3f) (Figure 8, Scheme 14) [92]. The same compound has also been produced more directly by reacting parabanic acid (24) with d6-benzene (43b) and triflic acid (Figure 8, Scheme 14) [93]. This work also prepared a number of deuterated derivatives of phenytoin, for example 3g. Phenytoin with deuteration of just one phenyl ring has been synthesised by Grignard production from d6-benzene (43b) and coupling with benzoylformaldehyde (46) to give d5-benzoin (37c) which was converted into d5-benzil (47) and then condensed with urea (26c) to give 5-(d5-phenyl)-5-phenylhydantoin (3h) (Figure 10, Scheme 15) [94]. This compound was used as a probe in metabolic studies and as an internal standard for combined GC-MS-computer analyses of body fluid extracts. The synthesis of the 3-hydroxyphenyl and 4-hydroxyphenyl metabolites of phenytoin labelled with deuterium in either of the two phenyl rings (48a, 48b, 49a, 49b) and of 5-(d5-phenyl)-5-(d4–p-hydroxyphenyl)-hydantoin (48c) (Figure 10) has been achieved by coupling the appropriate labelled and unlabelled components in the production of benzophenone followed by its use in a Bucherer-Bergs reaction (Figure 10, Scheme 16) [95].
Figure 10. Synthesis of deuterated and tritiated versions of phenytoin (5,5-diphenylhydantoin) (3) (Schemes 15-17).
The synthesis of phenytoin with a deuterium or tritium atom exclusively at the para position of both phenyl rings has been achieved by Bucherer-Bergs reaction with p-bromobenzophenone (50) to give 5,5-di-(4-bromophenyl)hydantoin (51) which was then reduced in the presence of deuterium oxide or tritium gas, replacing the bromines to give 5,5-di-(4-2H-phenyl)hydantoin (3i) [96] or 5,5-di-(4-3H-phenyl)-hydantoin (3j) [97], respectively (Figure 10, Scheme 17). In the case of the deuterated form, for materials with isotopic content up to 95%, excellent agreement was found between the data obtained by mass spectrometry and 13C-nuclear magnetic resonance operated in a NOE-suppression mode. Phenytoin has also been tritiated directly using 3H2O and platinum catalyst (from the dioxide and sodium borohydride) and the pattern of labelling was defined by 3H NMR spectroscopy [98].
An elegant seven-step synthesis has been used to prepare the separate enantiomers of phenytoin with a single deuterium atom at the ortho-position in only one of the phenyl groups (Figure 11, Scheme 18) [99]. The bromine atom of 3-bromoanisole (52) was substituted for deuterium by reduction with deuterium gas and the resultant [3-2H]methoxybenzene (53) was coupled with benzoyl chloride (35b) to give the ketone (54). Bucherer-Burgs reaction produced 5-([2-2H]4-methoxyphenyl)-5-phenylhydantoin (55) from which the methyl group was then removed. The enantiomers of 5-([2-2H]4-hydroxyphenyl)-5-phenylhydantoin (48e, 48f) were separated with brucine resolution [100] and then derivatised at the hydroxyl position with 5-chloro-1-phenyl-1H-tetrazole (56), which was removed under reduction conditions to give (R)-5-[2-2H]phenyl-5-phenylhydantoin (3k) and (S)-5-[2-2H]phenyl-5-phenylhydantoin (3l).
Figure 11. Synthesis of the separate enantiomers of 5-[2-2H]phenyl-5-phenylhydantoin (3k, 3l) (Scheme 18).
A synthesis of phenytoin with a deuterium atom at the ortho-position on both phenyl groups has also been achieved (Figure 12, Scheme 19) [101]. 2-Bromotoluene (58) was converted to the Grignard reagent and then decomposed with deuterium oxide to give [2-2H]toluene (59a) which was converted in three steps to [2-2H]benzaldehyde (36b). This was converted into the dideuterobenzoin (37d) and then dideuterobenzil which was rearranged to the dideuterobenzilic acid (60) before condensing with urea (26b) to give 5,5-di-[2-2H]phenylhydantoin (3m). A deuterium atom at the ortho-position in phenytoin is more stable to metabolism than at other positions and therefore more suitable to use in metabolic studies that are followed by mass spectrometry. A number of isotope-labelled derivatives of phenytoin with more complex substituents have also been synthesised. These include a series of 125I-labelled N3-substituted acetamido-tyrosine ethyl ester derivatives (61) used as iodinated tracers in a radioimmunoassay for phenytoin [102,103] and a [15N, 2H13]2,2,5,5-tetramethylpyrroline-1-oxyl-3-carboxylic acid derivative with the substituent at a phenyl para-position (62) also containing a spin label for detection by electron paramagnetic resonance (EPR) spectroscopy (Figure 12) [104].
![Figure 12 shows the synthesis of 5,5-di-[2-2H]phenylhydantoin (3m) (Scheme 19) and structures of phenytoin derivatives with 125I (61) and 15N,2H labels (62).](https://openmedscience.com/wp-content/uploads/2017/02/Figure-12-Synthesis-of-phenylhydantoin.jpg)
Figure 12. Synthesis of 5,5-di-[2-2H]phenylhydantoin (3m) (Scheme 19) and structures of phenytoin derivatives with 125I (61) and 15N,2H labels (62).
5. Other 5,5-disubstituted hydantoins
A number of other 5,5-disubstituted hydantoins have been synthesised and used in a range of different applications. A 13C-labelled benzylated hydantoin intermediate (66) has been used in the synthesis of the α-adrenergic agonist α-methyldopa with a 13C label at the benzylic position (67) (Figure 13, Scheme 20) [105]. A lithio derivative of 3,4-dibenzyloxybenzene (64) was subject to carbonation with [13C]carbon dioxide and the resultant benzoic acid (65) was reduced with lithium aluminium hydride. Oxidation with chromium trioxide gave the benzaldehyde which was condensed with nitroethane to form the phenylnitropropene. Reduction of the phenylnitropropene gave the corresponding phenyl-2-propanone which was converted to the hydantoin (66). The hydantoin was subject to base hydrolysis followed by debenzylation to give [13C]-α-methyldopa hydrochloride (67), which was used in biotransformation studies. The 5,5-cyclic disubstituted hydantoin sorbinil, an aldose reductase inhibitor with potential utility in the treatment of diabetes, has been prepared with a 14C label at C-4 in the hydantoin ring or with a deuterium or tritium atom at a single position on the aromatic ring of the substituent group (70a, 70b, 70c) (Figure 13, Scheme 21) [106].
Figure 13. Synthesis of other 5,5-disubstituted hydantoins.
A 13C-labelled precursor to α-methyldopa (66) (Scheme 20), 5,5-cyclic disubstituted sorbinil with 14C, deuterium or tritium labels (70a, 70b, 70c) (Scheme 21), deuterated and 14C-labelled versions of 5-ethyl-5-phenylhydantoin (71a,71b,71c), [2-13C, 1,3-15N2]spirodihydantoin (74a) (Scheme 22) and a series of 13C,15N-labelled hydantoin sulphonamide compounds (81) (Scheme 23).
The 14C label was introduced by Bucherer-Bergs reaction with the appropriate ketone (68) and [14C]potassium cyanide; deuterium or tritium was introduced by reductive dehalogenation of the chloride derivative (69) with deuterium or tritium gas. The pharmacologically active S-(+)-enantiomer was isolated by brucine resolution [100]. 5-Ethyl-5-phenylhydantoin has been synthesised with deuterium labelling at the methyl group (71a) or in the phenyl group (71b) or with a 14C-label at the C-4 position (71c) (Figure 13) during synthesis of the antimuscarinic drug trimebutine and its metabolites with deuterium and 14C labels [107]. The symmetric 5,5-cyclic disubstituted spirodihydantoin has been synthesised with 13C and 15N labels in one half of the molecule (74a) [108] using a method by Poje et al [109] (Figure 13, Scheme 22). Reaction of 4,5,6(1H)-pyrimidinetrione (72) with [13C,15N2]urea (26d) and then bromine gave the 5,5-disubstituted hydantoin (73) which was cyclised to give [2-13C, 1,3-15N2]spirodihydantoin (74a). This compound was used to help demonstrate that spirodihydantoin is a minor product from oxidation of uric acid. A series of 5,5-disubstituted hydantoins with a sulphonamide as one of the substituent groups have been prepared with multiple 13C labels and with a 15N label at N-3 in the hydantoin ring (81) using simple compounds as sources of the labels (Figure 13, Scheme 23) [110]. [13C3]Acetone (75) was converted into [13C3]chloroacetone (76), which was alkylated with benzyl mercaptan (77) then the ketone (78) was subjected to a microwave-induced Bucherer-Bergs reaction with [13C,15N]potassium cyanide to give the 5,5-disubstitued hydantoin (79), which was converted to the sulphonyl chloride (80). This was coupled with a range of amines to give the hydantoin sulphonamide compounds (81) with 13C labels at C-4, C-5, CH3 and CH2 and 15N at N-3. The introduction of multiple labels provided a molecule with a greater mass differential from the parent compound that was more useful than a molecule with a single label in the development of bioanalytical methods.
6. N-Substituted hydantoins
Some hydantoins with substituents at the N-1 and/or N-3 position have been synthesised with isotope labels. The hydantoin drug dantrolene (82), which interferes with release of calcium ions from intracellular stores of skeletal muscle, and a photoaffinity azido analogue (83) have been synthesised with a tritium label at a non-exchangeable imine in the N-1 substituent group (Figure 14) [111].
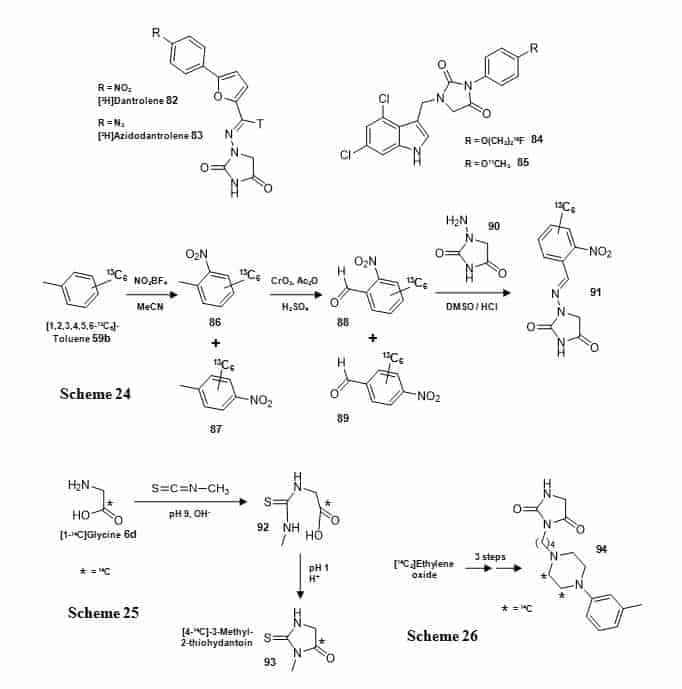
Figure 14. N–Substituted hydantoins.
Structures of [3H]dantrolene (82), [3H]azidodantrolene (83), and 19F- or 11C-labelled NMDA receptor compounds (84, 85), and syntheses of a 13C-labelled 2-nitrobenzaldehyde derivative of 1-aminohydantoin (91) (Scheme 24), [4-14C]-3-methyl-2-thiohydantoin (93) (Scheme 25) and a 14C-labelled antihypertensive agent (94) (Scheme 26).
The tritium label was introduced into [3H]dantrolene using sodium borotritide and into [3H]azidodantrolene using lithium triethylborotritide. 1H and 3H NMR analysis of the final tritiated compounds suggested ≥95% 3H labelling since the spectra showed no sign of hydrogen at the imine position (Figure 15A). It is also reported that the compounds showed no sign of hydrogen/tritium exchange during a period of over one year [111]. The tritiated compounds were used in the identification of a putative skeletal muscle dantrolene binding site in sarcoplasmic reticulum membranes (Figure 15B) [111]. Dantrolene is also a substrate for breast cancer resistant protein (BCRP), which is widely distributed in the blood-brain-barrier, intestine, gall bladder and liver.
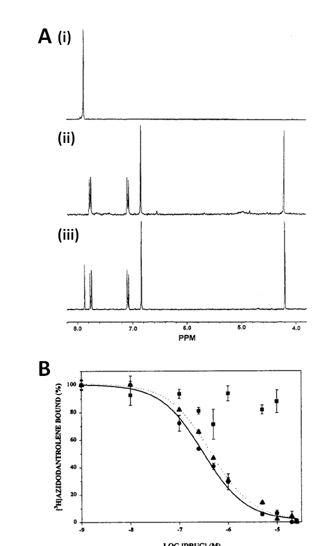
Figure 15. (Figures 13 and 14 are on pages 22 and 23).
NMR analysis of [3H]dantrolene (82) and [3H]azidodantrolene (83) and use of [3H]azidodantrolene in identifying a dantrolene binding site in sarcoplasmic reticulum membranes. A. (i) 3H NMR spectrum of [3H]azidodantrolene in d8-THF, (ii) 1H NMR spectrum of [3H]azidodantrolene in d8-THF and (iii) 1H NMR spectrum of [3H]dantrolene in d8-THF. B. Inhibition of [3H]azidodantrolene (200 nM) binding to sarcoplasmic reticulum membranes by increasing concentrations of dantrolene (triangles), azumolene (circles) or aminodantrolene (squares). Dantrolene and azumolene were equipotent whilst aminodantrolene was inactive. These figures were reproduced with permission from Palnitkar et al (1999) [111]; copyright © 1999 by the American Chemical Society.
Radiolabelled versions of dantrolene have been synthesised for use in positron emission tomography (PET) imaging studies that target BCRP. [2-11C-carbonyl]Dantrolene has been prepared in a multi-step/one-pot labelling sequence starting with ethyl 2-{2-[5-(4-nitrophenyl)furfurylidene]hydrazino} acetate and using [11C]phosgene as the labelling agent [112]. [13N]Dantrolene has been prepared using no-carrier-added [13N]ammonia as the labelling agent [113]. A number of hydantoin‐substituted indole‐2‐carboxylic acid compounds targeted at the N‐methyl‐D‐aspartate (NMDA) receptor have been synthesised, which included two reference compounds with a fluoroethoxy or methoxy group labelled with 18F or 11C (84, 85) (Figure 14), respectively, for PET imaging of the receptor glycine binding site [114]. These compounds containing a 1,3-di-N-substituted hydantoin moiety were obtained by alkylation of a phenolic hydroxyl group using [18F]-2-fluoroethyl tosylate or [11C]methyl iodide. A 2-nitrobenzaldehyde derivative of 1-aminohydantoin (91) has been synthesised with 13C labels in the aromatic ring starting from [1,2,3,4,5,6-13C6]toluene (59b) (Figure 14, Scheme 24) [115]. The [13C6]toluene (59b) was nitrated using nitronium tetrafluoroborate giving a mixture of ortho and para products (86, 87) which were then simultaneously oxidised using chromium trioxide to give a mixture of [1,2,3,4,5,6-13C6]-2-nitrobenzaldehyde (88) and [1,2,3,4,5,6-13C6]-4-nitrobenzaldehyde (89). Separation on a silica gel column using a step gradient of dichloromethane afforded the required ortho isomer in a yield of not more than 10% from [13C6]toluene, which was then coupled with 1-aminohydantoin (90) to give (91). A full 1H and 13C NMR analysis of the final labelled compound was performed with chemical shift assignments assisted by [1H-1H]COSY, [1H-13C]HMQC and [1H-13C]HMBC 2D-correlation spectra and a homonuclear NOE difference spectrum exhibited a spatial correlation between the iminyl hydrogen and the hydrogen atoms N-CH2 indicative of an E configuration [115]. This compound was used along with other 13C-labelled 2-nitrobenzaldehyde derivatives of nitrofuran metabolites as internal standards for the quantification of trace levels of nitrofuran residues by liquid chromatography-tandem mass spectrometry in foods of animal origin [115]. 3-Methyl-2-thiohydantoin (93) has been synthesised with a 14C label at C-4 in two steps from [1-14C]glycine (6d) (Figure 14, Scheme 25) [116]. Reaction of the glycine with methyl isothiocyanate under basic conditions produced 14C-labelled 5-methylthio-hydantoic acid (92), which cyclises at acidic pH to [4-14C]-3-methyl-2-thiohydantoin (93). This compound is a pharmacologically active metabolite of the antithyroid drug methimazole. A new antihypertensive agent 3-[4-]4-(3-methylphenyl)-1-piperazinyl[butyl]hydantoin (94) was synthesised with two adjacent 14C labels in the piperazinyl ring in three steps from [14C2]ethylene oxide with an overall radiochemical yield of 69% (Figure 14, Scheme 26) [117].
7. 18O-Labelled hydantoins
The stable isotope oxygen-18 has been incorporated into some hydantoin compounds. Guanidinohydantoin has been synthesised with 13C, 15N and 18O labels (96) by reaction of 13C,15N-labelled guanosine (95) with singlet oxygen and [18O]water at a pH of less than 7 (Figure 16, Scheme 27) [118]. This isotope labelling of guanidinohydantoin combined with mass spectrometry and NMR analysis helped to resolve controversial issues concerning its structure and suggested potential mechanisms to explain products observed from the oxidation of guanosine by singlet oxygen, which has importance in cancer etiology and its cure by photodynamic therapy. The same work that produced 13C,15N-labelled spirodihydantoin (74a) described above also produced spirodihydantoin (74b), spiroiminohydantoin (97), allantoin (2b) and guanidinohydantoin (96) with an 18O label (Figure 16) [108]. These compounds were used in an investigation of uric acid metabolism.
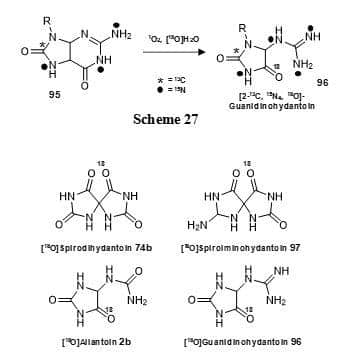
Figure 16. 18O-labelled hydantoins.
Synthesis of guanidinohydantoin with 13C, 15N and 18O labels (96) (Scheme 27) and structures of 18O-labelled spirodihydantoin (74b), spiroiminohydantoin (97), allantoin (2b) and guanidinohydantoin (96).
8. Other more complex molecules that contain the hydantoin moiety
A range of compounds containing the hydantoin moiety with multiple and/or complex substituents have been synthesised containing isotope labels. The separate enantiomers of the anticonvulsant drug 3-methyl-5-ethyl-5-phenylhydantoin (mephenytoin) (98) were synthesised with a 14C label at the 3-methyl group by reaction of the separate R and S forms of 5-ethyl-5-phenylhydantoin (71) with [14C2]dimethylsulphate (Figure 17, Scheme 28) [119]. These compounds were used to measure their fate of metabolism in breath, blood and urine of the dog. The nitrogen mustard spiromustine (101) has been synthesised in an octadeuterated form (Figure 17, Scheme 29) [120]. d6-Ethylene glycol was converted d8-ethanolamine via a 2-imino-1,3-oxazolidine intermediate. The d8-ethanolamine was then reacted with 3-chloroethyl-spirohydantoin (99) to give d8-spiromustine (101) and other deuterated analogues. At the time, spiromustine was a potential new antitumor agent designed specifically for neoplasms of the central nervous system and the deuterated form was required as an internal standard for selected ion monitoring by GC-MS.
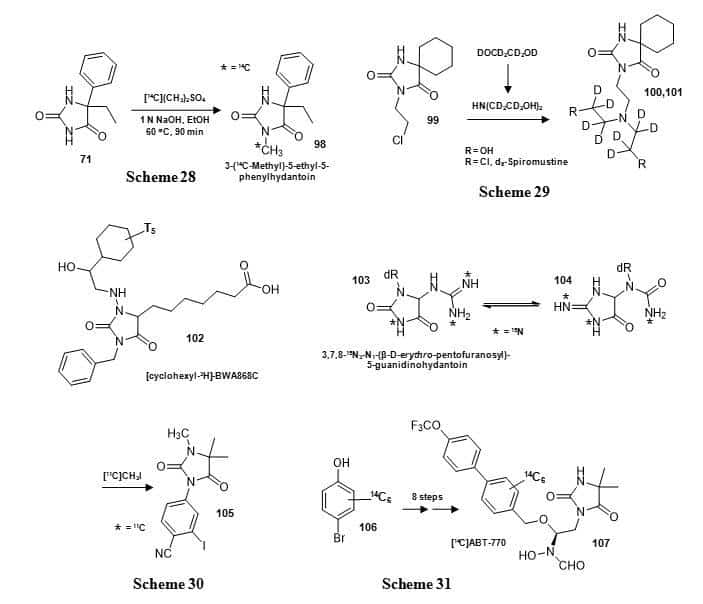
Figure 17. Syntheses of 14C-labelled mephenytoin (98) (Scheme 28) and d8-spiromustine (101) (Scheme 29), structures of a tritium labelled DP prostanoid receptor antagonist BWA868C (102) and 15N3-labelled N1-(β-D-erythro-pentofuranosyl)-5-guanidinohydantoin (103) and syntheses of an 11C-labelled nonsteroidal androgen receptor ligand (105) (Scheme 30) and a 14C-labelled matrix metalloproteinase inhibitor ABT-770 (107) (Scheme 31).
A DP prostanoid receptor antagonist BWA868C or 3-benzyl-5-(6-carboxyhexyl)-1-(2-cyclohexyl-2-hydroxy-ethyl-amino)hydantoin (102) has been synthesised with tritium labelling in the cyclohexyl group (Figure 17) and then used for characterisation and autoradiographic localisation of DP receptors in a range of mammalian tissues and cells and to better define the distribution and possible functions of DP receptors in the mammalian body [121]. This includes localisation of DP receptors in the human eye (Figure 18) [121].
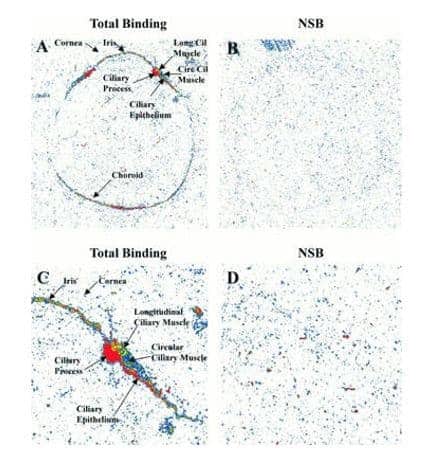
Figure 18. Localisation of DP prostanoid receptors in the human eye using antagonist [3H]BWA868C.
Quantitative autoradiographs of DP prostanoid receptor binding sites (highest intensity = red) in sections of human eye using 5 nM [3H]BWA868C. Panels A and C show total binding, B and D show non-specific binding (NSB). Magnifications for the images are 3.4x (A and B) and 8.5x (C and D). This picture was reproduced with permission from Sharif et al (2000) [121]; copyright © 2000 by the British Pharmacological Society.
A 15N3-labelled form of N1-(β-D–erythro-pentofuranosyl)-5-guanidinohydantoin (103) has been synthesised from [1-15N,7-15N,15NH2]2′-deoxyguanosine for use as an internal standard in the measurement of N1-(β-D–erythro-pentofuranosyl)-5-guanidinohydantoin in peroxynitrite oxidized DNA by isotope-dilution mass spectrometry [122]. Compound 103 undergoes pH- and temperature-dependent isomerisation to the iminoallantoin (104) (Figure 17), which made its purification especially challenging.
A multi-substituted hydantoin nonsteroidal androgen receptor ligand (105) was synthesised with an 11C label by introduction of the N-1 methyl group in the final step using [11C]methyl iodide (Figure 17, Scheme 30) [123]. This compound was used as a potential radioligand for PET imaging of prostate cancer. A matrix metalloproteinase inhibitor ABT-770: N-[(1S)-1-[(4,4-dimethyl-2,5-dioxo-1-imidazolidinyl)methyl]]-2-[[40-(trifluoromethoxy)[1,10-biphenyl]-4-yl]oxy]ethyl]-N-hydroxy formamide (107) was synthesised in 8 steps from [14C6]4-bromophenol (106), which introduces 14C-labelling into a metabolically stable biphenyl ring (Figure 17, Scheme 31) [124]. The key part of the synthesis was a three-step one-pot reaction in which the hydantoin moiety was introduced, the imine oxidized and further hydrolysed to obtain the penultimate precursor to [14C]ABT-770 (107). The compound was used in animal metabolism studies as a potential new antitumor agent. The synthesis of conformationally constrained analogues of glutamic acid: (+)-2-aminobicyclo[3.1.0]hexane-2,6-carboxylic acid and its 2-oxa and 2-thia-analogues with general structure 109 containing 3H, 13C, 14C and 15N labels included a key hydantoin intermediate, for example 108, which was formed by Bucherer-Burgs reaction with the appropriate chiral ketone and then hydrolysed to give 109 (Figure 19) [125].
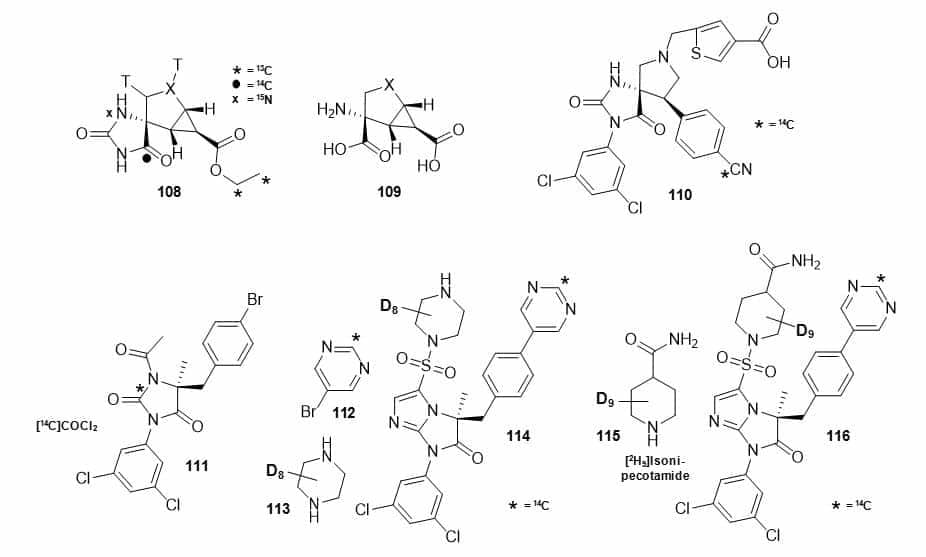
Figure 19. Structures of a hydantoin intermediate with 3H, 13C, 14C and 15N labels (108) used as a precursor to conformationally constrained analogues of glutamic acid and 2-oxa and 2-thia-analogues (109), a 14C-labelled spyrocyclic hydantoin (110), and antagonists of lymphocyte function-associated antigen-1 with 14C and/or deuterium labels (111, 114, 116).
Isotope labels were introduced using tritium gas, [13C2]ethyl bromoacetate, [13C]potassium cyanide, [14C]potassium cyanide and [15N]ammonium chloride. This work also described the first use of in situ generated [15N]ammonium carbonate in the Bucherer-Bergs reaction, which was formed by reaction of [15N]ammonium chloride and sodium carbonate. These compounds had been identified as highky potent glutamate receptor agonists and isotope-labelled versions were synthesised for preclinical ADME studies. The drug lead candidate leukocyte function-associated antigen 1 antagonist spyrocyclic hydantoin: 5-(((5S,9R)-9-(4-cyanophenyl)-3-(3,5-dichloro-phenyl)-1-methyl-2,4-dioxo-1,3,7-triazaspiro[4.4]nonan-7-yl)methyl)-thiophene-3-carboxylic acid (110) (Figure 19) was synthesised with a 14C label at the cyano group by substitution of a bromide with [14C]zinc cyanide, prepared from zinc chloride and [14C]potassium cyanide, followed by two further radiochemical steps [126]. Three hydantoin based potent antagonists of lymphocyte function-associated antigen-1 labelled with 14C and deuterium were synthesised for drug metabolism and pharmacokinetics studies (Figure 19) [127]. (R)-1-acetyl-5-(4-bromobenzyl)-3-(3,5-dichloro-phenyl)-5-methyl hydantoin (111) was prepared with a 14C label at C-2 in the hydantoin ring in two radiochemical steps using [14C]phosgene. (R)-5-(1-piperazinylsulfonyl)-1-(3,5-dichlorophenyl)-3-[4-(5-pyrimidinyl)benzyl]-3-methyl-1-H-imidazo[1,2a]imidazol-2-one (114) and (R)-1-[7-(3,5-dichlorophenyl)-5-methyl-6-oxo-5-(4-pyrimidin-5-ylbenzyl)-6,7-dihydro-5H-imidazo[1,2-a]imidazole-3-sulfonyl]piperidin-4-carboxylic acid amide (116) were prepared with a 14C label in the pyrimidine ring using [2-14C]-5-bromopyrimidine (112) in a Suzuki reaction with the appropriate boronic acid esters.
A deuterated version of 114 was prepared by reaction of the sulphonyl chloride derivative with d8-piperazine (113) and a deuterated version of 116 was prepared by reaction of the sulphonyl chloride derivative with d9-isonipecotamide (115). A 125I-labelled thiohydantoin derivative (117) (Figure 20) has been synthesised using [125I]sodium iodide as the source of the label in the last step via an iododestannylation reaction with hydrogen peroxide as the oxidant and used for detecting tau pathology in Alzheimer’s disease [128]. In vitro experiments using tau and β-amyloid aggregates showed high specific binding of 117 to tau aggregates and in hippocampal sections of brains from Alzheimer’s disease patients, 117 produced intense staining of neurofibrillary tangles (Figure 20). Furthermore, in experiments using normal mice, 117 showed good uptake into and rapid washout from the brain, suggesting that a 123I-labelled version of 117 should be investigated as a PET radiotracer for imaging tau pathology [128].
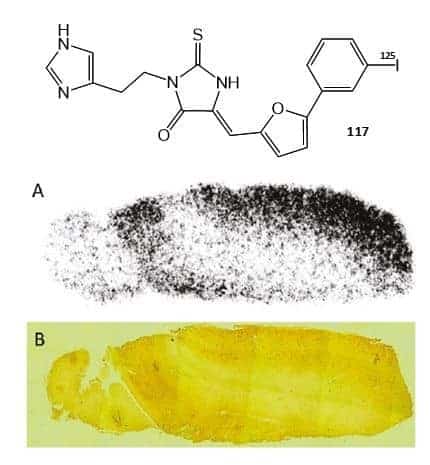
Figure 20. Structure of a 125I-labelled thiohydantoin derivative (117) and its use for detecting tau pathology in Alzheimer’s disease.
A. In vitro autoradiogram of a section of human Alzheimer’s disease brain labelled with 117. B. The same section of Alzheimer’s disease brain immunostained with an antibody against hyperphosphorylated tau. The pictures in A and B were reproduced with permission from Ono et al (2011) [128]; copyright © 2011 by the American Chemical Society.
9. Conclusions
This review of the methods of synthesis, NMR analysis and applications of isotope-labelled hydantoins has confirmed the hydantoin ring system as an important centre in synthetic chemistry and as a component in a variety of different compounds including some with pharmacological activity. In some cases, NMR analysis of the labelled compounds was especially important for confirming purity, labelling integrity and molecule conformation. Hydantoin compounds containing isotope labels have been used in a range of biological, biomedical, food and environmental applications including metabolic and in vivo tissue distribution studies, biochemical analysis of transport proteins, identification and tissue distribution of drug binding sites, drug metabolism and pharmacokinetic studies and as an imaging agent.
Conflict of interest
The author confirms that this article content has no conflict of interest.
Funding
This work was supported by the EU EDICT consortium (contract 201924) and the BBSRC through grant numbers BB/C51725X/1 and BB/G020043/1.
References
Key references: 58, 59, 60, 62, 64, 71, 85, 90, 111, 128
- Pettit GR, Herald CL, Leet JE, et al. Antineoplastic agents. 168. Isolation and structure of axinohyd antoin. Can J Chem. 1990; 68(9): 1621-1624. CrossRef
- Mudit M, Behery FA, Wali VB, Sylvester PW, El Sayed KA. Synthesis of fluorescent analogues of the anticancer natural products 4-hydroxyphenylmethylene hydantoin and d-tocotrienol. Nat Prod Commun. 2010; 5(10): 1623-1626. PubMed
- Cachet N, Genta-Jouve G, Regalado EL, Mokrini R, Amade P, Culioli G, Thomas OP. Parazoanthines A-E, hydantoin alkaloids from the Mediterranean sea anemone Parazoanthus axinellae. J Nat Prod. 2009; 72(9): 1612-1615. PubMed
- Wen H, Li Y, Liu X, Ye W, Yao X, Che Y. Fusagerins A-F, New alkaloids from the fungus Fusarium sp. Nat Prod Bioprospect. 2015; 5(4): 195-203. CrossRef PubMed
- Fang X, Mao J, Cory RM, McKnight DM, Schmidt-Rohr K. 15N and 13C{14N} NMR investigation of the major nitrogen-containing segment in an aquatic fulvic acid: Evidence for a hydantoin derivative. Magn Reson Chem. 2011; 49(12): 775-780. CrossRef
- de Marcellus P, Bertrand M, Nuevo M, Westall F, d’Hendecourt LLS. Prebiotic significance of extraterrestrial ice photochemistry: Detection of hydantoin in organic residues. Astrobiology. 2011; 11(9): 847-854. CrossRef PubMed
- Menor-Salván C, Marín-Yaseli MR. A new route for the prebiotic synthesis of nucleobases and hydantoins in water/ice solutions involving the photochemistry of acetylene. Chemistry. 2013; 19(20): 6488-6497. PubMed
- von Baeyer A. Mittheilungen aus dem organischen laboratorium des gewerbeinstitutes in Berlin: Untersuchungen über die harnsäuregruppe. Ann Chem Pharm. 1864; 130(2): 129-175.Reference Source
- Strecker A. Ueber uroxansäure und die chemische constitution der harnsäure. Ann Chem. 1870; 155(2): 177-185. Reference Source
- Biltz H, Slotta K. Ueber die herstellung von hydantoinen. J Prakt Chem. 1926; 113:233-267. Reference Source
- Ware E. The chemistry of the hydantoins. Chem Rev. 1950; 46(3): 403-470. CrossRef PubMed
- Schipper ES, Day AR. In Heterocyclic Compounds (Elderfield RC, Ed.), Vol. 5, John Wiley & Sons Inc., New York, 1957, p. 254. Reference Source
- López CA, Trigo GG. The chemistry of hydantoins. Adv Heterocycl Chem. 1985;38: 177-228. CrossRef
- Avendaño C, Menendez JC. Hydantoin and its derivatives. In Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc, 2000. Reference Source
- Meusela M, Gütschowa M. Recent developments in hydantoin chemistry. A review. Org Prep Proced Int. 2004; 36(5): 391-443. CrossRef
- Boeijen A, Kruijtzer JAW, Liskamp RMJ. Combinatorial chemistry of hydantoins. Bioorg Med Chem Lett. 1998; 8(17): 2375-2380. CrossRef PubMed
- Olimpieri F, Bellucci MC, Marcelli T, Volonterio A. Regioselective multicomponent sequential synthesis of hydantoins. Org Biomol Chem. 2012; 10(48): 9538-9555. CrossRef PubMed
- Belluccia MC, Frigeriob M, Marcelli T, Volonterio A. Multicomponent synthesis of N-carbamoyl hydantoin derivatives. Syn Lett. 2013;24(6): 727-732. Reference Source
- Zhang W, Lu Y, Hiu-Tung CC, Zeng L, Kassel DB. Fluorous mixture synthesis of two libraries with novel hydantoin- and benzodiazepinedione-fused heterocyclic scaffolds. J Comb Chem. 2006;8(5): 687-695. PubMed
- Thomas GL, Wyatt EE, Spring DR. Enriching chemical space with diversity-oriented synthesis. Curr Opin Drug Discov Dev. 2006; 9(6): 700-712. CrossRef
- Dhara K, Midya GC, Dash J. A diversity-oriented approach to spirocyclic and fused hydantoins via olefin metathesis. J Org Chem. 2012;77(18): 8071-8082. CrossRef PubMed
- Olivieri R, Fascetti E, Angelini L, Degen L. Microbial transformation of racemic hydantoins to D-amino acids. Biotechnol Bioeng. 1981; 23(10): 2173-2183. CrossRef
- Rai R, Rao RB, Taneja V. Hydrolysis of di-substituted hydantoins, by an enzyme preparation from lentil (Lens esculenta) seeds, for the synthesis of α,α-dialkylated amino acids with linear and cyclic substituents. World J Microb Biot. 1996; 12(3): 247-250. CrossRef PubMed
- Durham DR, Weber JE. Stereospecific preparation of an excitatory amino acid antagonist with D-hydantoinase from Agrobacterium tumefaciens as a biocatalyst. Appl Environ Microb. 1996; 62(2): 739-742. PubMed
- Ishikawa T, Watabe K, Mukohara Y, Nakamura H. Mechanism of stereospecific conversion of dl-5-substituted hydantoins to the corresponding l-amino acids by Pseudomonas sp. strain NS671. Biosci Biotechnol Biochem. 1997; 61(1): 185-187. PubMed
- Syldatk C, May O, Altenbuchner J, Mattes R, Siemann M. Microbial hydantoinases – industrial enzymes from the origin of life? Appl Microbiol Biotechnol. 1999;51(3): 293-309. CrossRef PubMed
- Altenbuchner J, Siemann-Herzberg M, Syldatk C. Hydantoinases and related enzymes as biocatalysts for the synthesis of unnatural chiral amino acids. Curr Opin Biotechnol. 2001; 12(6): 559-563. CrossRef PubMed
- Wilms B, Wiese A, Syldatk C, Mattes R, Altenbuchner J. Development of an Escherichia coli whole cell biocatalyst for the production of L-amino acids J. Biotechnol. 2001; 86(1): 19-30. CrossRef PubMed
- Martinez-Rodriguez S, Las Heras-Vazquez FJ, Clemente-Jimenez JM, et al. Complete conversion of D,L-5-monosubstituted hydantoins with a low velocity of chemical racemization into D-amino acids using whole cells of recombinant Escherichia coli. Biotechnol Prog. 2002; 18(6): 1201-1206. CrossRef PubMed
- Clemente-Jiménez JM, Martínez-Rodríguez S, Rodríguez-Vico F, Heras-Vázquez FJ. Optically pure α-amino acids production by the “Hydantoinase Process” Recent Pat Biotechnol. 2008; 2(1): 35-46. PubMed
- Nandanwar HS, Hoondal GS, Vohra RM. Enzymatic production of D-amino acids. In Methods in Biotechnology (Barredo JL, Ed.), 2005, Vol. 17: Microbial enzymes and biotransformations, Humana Press, p. 91-104. Reference Source
- Martínez-Gómez AI, Martínez-Rodríguez S, Clemente-Jiménez JM, Pozo-Dengra J, Rodríguez-Vico F, Las Heras-Vázquez FJ. Recombinant polycistronic structure of hydantoinase process genes in Escherichia coli for the production of optically pure D-amino acids. Appl Environ Microbiol. 2007; 73(5): 1525-1531. PubMed
- Martínez-Rodríguez S, Martínez-Gómez AI, Rodríguez-Vico F, Clemente-Jiménez JM, Las Heras-Vázquez FJ. Natural occurrence and industrial applications of D-amino acids: an overview. Chem Biodivers. 2010; 7(6): 1531-1548. CrossRef PubMed
- Mehta NB, Diuguid CA, Soroko FE. Potential anticonvulsants. 1. 5-Benzylhydantoins. J Med Chem. 1981; 24(4): 465-468. CrossRef PubMed
- Poupaert JH, Vandervorst D, Guiot P, Moustafa MM, Dumont P. Structure-activity relationships of phenytoin-like anticonvulsant drugs. J Med Chem. 1984; 27(1): 76-78. CrossRef PubMed
- Mishory A, Yaroslavsky Y, Bersudsky Y, Belmaker RH. Phenytoin as an antimanic anticonvulsant: a controlled study. Am J Psychiatry. 2000; 157(3): 463-465. CrossRef PubMed
- Lenkowski PW, Batts TW, Smith MD, et al. A pharmacophore derived phenytoin analogue with increased affinity for slow inactivated sodium channels exhibits a desired anticonvulsant profile. Neuropharmacology. 2007; 52(3): 1044-1054. CrossRef PubMed
- Suzuki S, Takenaka Y, Onishi N, Yokozeki K. Molecular cloning and expression of the hyu genes from Microbacterium liquefaciens AJ 3912, responsible for the conversion of 5-substituted hydantoins to α-amino acids, in Escherichia coli. Biosci Biotechnol Biochem. 2005; 69(8): 1473-1482. PubMed
- Suzuki S, Henderson PJ. The hydantoin transport protein from Microbacterium liquefaciens. J Bacteriol. 2006; 188(9): 3329-3336. PubMed
- Weyand S, Shimamura T, Yajima S, et al. Structure and molecular mechanism of a nucleobase-cation-symport-1 family transporter. Science. 2008; 322(5902): 709-713. CrossRef PubMed
- Shimamura T, Weyand S, Beckstein O, et al. Molecular basis of alternating access membrane transport by the sodium-hydantoin transporter Mhp1. Science. 2010; 328(5977): 470-473. CrossRef PubMed
- Weyand S, Shimamura T, Beckstein O, Sansom MS, Iwata S, Henderson PJ, Cameron AD. The alternating access mechanism of transport as observed in the sodium-hydantoin transporter Mhp1. J Synchrotron Radiat. 2011; 18(1): 20-23. CrossRef PubMed
- Jackson SM, Patching SG, Ivanova E, et al. Mhp1, the Na+-hydantoin membrane transport protein. In Encyclopedia of Biophysics (Roberts GCK, Ed.), 2013, Springer, p. 1514-1521. Reference Source
- de Koning H, Diallinas G. Nucleobase transporters (review). Mol Membr Biol. 2000; 17(2): 75-94. PubMed
- Pantazopoulou A, Diallinas G. Fungal nucleobase transporters. FEMS Microbiol Rev. 2007; 31(6): 657-675. CrossRef PubMed
- Weyand S, Ma P, Saidijam M, et al. The nucleobase-cation-symport-1 family of membrane transport proteins. In Handbook of Metalloproteins, 2010, part 11, John Wiley and Sons. Reference Source
- Hamari Z, Amillis S, Drevet C, Apostolaki A, Vágvölgyi C, Diallinas G, Scazzocchio C. Convergent evolution and orphan genes in the Fur4p-like family and characterization of a general nucleoside transporter in Aspergillus nidulans. Mol Microbiol. 2009; 73(1): 43-57. PubMed
- Krypotou E, Evangelidis T, Bobonis J, et al. Origin, diversification and substrate specificity in the family of NCS1/FUR transporters. Mol Microbiol. 2015; 96(5): 927-950. CrossRef PubMed
- Mourad GS, Tippmann-Crosby J, Hunt KA, Gicheru Y, Bade K, Mansfield TA, Schultes NP. Genetic and molecular characterization reveals a unique nucleobase cation symporter 1 in Arabidopsis. FEBS Lett. 2012, 586(9): 1370-1378. CrossRef PubMed
- Witz S, Panwar P, Schober M, Deppe J, Pasha FA, Lemieux MJ, Möhlmann T. Structure-function relationship of a plant NCS1 member – homology modeling and mutagenesis identified residues critical for substrate specificity of PLUTO, a nucleobase transporter from Arabidopsis. PLoS One. 2014; 9(3): e91343. CrossRef PubMed
- Rapp M, Schein J, Hunt KA, Nalam V, Mourad GS, Schultes NP. The solute specificity profiles of nucleobase cation symporter 1 (NCS1) from Zea mays and Setaria viridis illustrate functional flexibility. Protoplasma. 2016; 253(2): 611-623. CrossRef PubMed
- Minton JA, Rapp M, Stoffer AJ, Schultes NP, Mourad GS. Heterologous complementation studies reveal the solute transport profiles of a two-member nucleobase cation symporter 1 (NCS1) family in Physcomitrella patens. Plant Physiol Biochem. 2016; 100: 12-17. CrossRef PubMed
- Adelman JL, Dale AL, Zwier MC, Bhatt D, Chong LT, Zuckerman DM, Grabe M. Simulations of the alternating access mechanism of the sodium symporter Mhp1. Biophys J. 2011;101(10): 2399-2407. CrossRef PubMed
- Shi Y. Common folds and transport mechanisms of secondary active transporters. Annu Rev Biophys. 2013; 42: 51-72. CrossRef PubMed
- Zhao C, Noskov SY. The molecular mechanism of ion-dependent gating in secondary transporters. PLoS Comput Biol. 2013; 9(10): e1003296. CrossRef PubMed
- Kazmier K, Sharma S, Islam SM, Roux B, Mchaourab HS. Conformational cycle and ion-coupling mechanism of the Na+/hydantoin transporter Mhp1. Proc Natl Acad Sci U S A. 2014; 111(41): 14752-14757. CrossRef PubMed
- Song HD, Zhu F. Conformational changes in two inter-helical loops of Mhp1 membrane transporter. PLoS One. 2015; 10(7): e0133388. CrossRef PubMed
- Ma P, Patching SG, Ivanova E, Baldwin JM, Sharples D, Baldwin SA, Henderson PJ. Allantoin transport protein, PucI, from Bacillus subtilis: evolutionary relationships, amplified expression, activity and specificity. Microbiology. 2016; 162(5): 823-836. CrossRef PubMed
- Patching SG. Synthesis of highly pure 14C-labelled DL-allantoin and 13C NMR analysis of labelling integrity. J Label Compd Radiopharm. 2009; 52(9): 401-404. Reference Source
- Patching SG. Efficient syntheses of 13C- and 14C-labelled 5-benzyl and 5-indolylmethyl L-hydantoins. J Label Compd Radiopharm. 2011; 54(2): 110-114. CrossRef
- Patching SG, Jackson S, Simmons KJ, et al. Isotope chemistry for investigating ligand binding and transport by Mhp1 and its homologues. J Label Compd Radiopharm. 2012; 55: 132. Reference Source
- Simmons KJ, Jackson SM, Brueckner F, Patching SG, et al. Molecular mechanism of ligand recognition by membrane transport protein, Mhp1. EMBO J. 2014;33(16): 1831-1844. CrossRef PubMed
- Herráez A. Biomolecules in the computer: Jmol to the rescue. Biochem Mol Biol Educ. 2006; 34(4): 255-261. PubMed
- Bond HW. Synthesis of carboxyl-labeled tryptophan from hydantoin containing isotopic carbon. J Biol Chem. 1948; 175(2): 531-534. PubMed
- Tolman V, Hanuš J, Vereš K. Synthesis of 14C-labelled heterocyclic analogues of phenylalanine with pyrazole and furan rings. J Label Compd. 1968;4(3): 243-247. CrossRef
- Tang G, Tang X, Wang S, Jiang G, Wu S. Synthesis of DL-α-15N-tryptophan from 1-15N-hydantoin. J Label Compd Radiopharm. 1999;42(2):199-201. CrossRef
- Bergs H, Ger. Pat. 566,094 (1929). Reference Source
- Bucherer HT, Fischbeck HT. Hexahydrodiphenylamine and its derivatives. J Prakt Chem. 1934; 140: 69-89.
- Bucherer HT, Steiner W. Uber die reaktionen der α-oxy und α-aminonitrilen. Synthesc von hydantoinen. J Prakt Chem. 1934; 140: 291-316.
- Shirai T, Takei Y. Studies on the products from formaldehyde by the Bucherer-Bergs synthesis: Studies on amino acids. I. J Syn Org Chem. 1971; 29(12):1135-1141. CrossRef
- Winstead MB, Parr SJ, Rogal MJ, et al. Relationship of molecular structure to in vivo scintigraphic distribution patterns of carbon-11 labeled compounds. 3. (11C)Hydantoins. J Med Chem. 1976; 19(2): 279-286. PubMed
- Kim JH, Creger CR, Couch JR. Synthesis of DL-tyrosine-4-14C. J Label Compd. 1969; 5(1): 35-41. CrossRef
- Reid JC. Synthesis of tyrosine labeled with C14. Science. 1947; 105(2721): 208. CrossRef PubMed
- Reid JC, Jones HB. Radioactivity distribution in the tissues of mice bearing melanosarcoma after administration of DL-tyrosine labeled with radioactive carbon. J Biol Chem. 1948; 174(2): 427-437. PubMed
- Loftfield RB. Synthesis of C-14 carboxyl-labeled tyrosine and diiodotyrosine. J Am Chem Soc. 1950; 72(6): 2499-2500. Reference Source
- Clemo GR, Duxbury FK, Swan GA. 664. Formation of tyrosine melanin. Part III. The use of carboxyl-labelled tyrosine and dihydroxyphenylalanine in melanin formation. J Chem Soc. 1952: 3464-3468. CrossRef
- Ramamurthy TV, Pichat L. Preparation of high specific activity DL-tryptophan-2, 3-T and its resolution to l-tryptophan-2, 3-T. J Label Compd. 1973; 9(2): 325-330. CrossRef
- Gaudry R. The synthesis of D,L-α-amino-e-hydroxycaproic acid and a new synthesis of D,L-lysine. Can J Res. 1948; 26(4 Sect B): 387-392. PubMed
- Borsook H, Deasy CL, Haagen-Smit AJ, Keighly G, Lowy PH. The uptake in vitro of C14-labeled glycine, l-leucine, and l-lysine by different components of guinea pig liver homogenate. J Biol Chem. 1950; 184(2): 529-543. PubMed
- Knapp FF Jr, Ambrose KR, Callahan AP. Potential pancreatic imaging agents. Tellurium-123m labeled DL-α-amino-g-(phenyltelluro)butyric acid. J Med Chem. 1981; 24(7): 794-797. CrossRef PubMed
- Abblard J, Meynaud A. Structure of allantoin. Bull Soc Chim Fr. 1971; 3: 942-946.
- Urech F. XXI. Ueber lacturaminsäure und lactylharnstoff. Ann Chem. 1873; 165(1): 99-103. Reference Source
- Rosenthal GA, Thomas D. Radiochemical synthesis of DL-canaline and the colorimetric assay of canaline. Anal Biochem. 1984; 140(1): 246-249. CrossRef PubMed
- Kepler JA, Lytle JW, Taylor GF. Synthesis of 5,5-diphenylhydantion-2,4,5-13C3. J Label Compd. 1974; 10(4): 683-687. CrossRef
- Emran AM, Boothe TE, Finn RD, Vora MM, Kothari PJ. Use of 11C as a tracer for studying the synthesis of radiolabelled compounds-II: 2-[11C]-5,5-diphenylhydantoin from [11C]cyanide. Int J Radiation Applications Instrumentation, Part A, Applied Radiation & Isotopes. 1986; 37(10): 1033-1038. CrossRef
- Iida K, Chiyoda T, Hirasawa R, Iwata A, Kajiwara M. Synthesis of 13C-labelled compounds having a urea unit, and observation of 13C-isotope effect in their infrared spectra. J Label Compd Radiopharm. 1997; 39(1): 69-77. CrossRef
- Roeda D, Westera G. The synthesis of some 11C-labelled antiepileptic drugs with potential utility as radiopharmaceuticals: hydantoins and barbiturates. Int J Appl Radiat Isot. 1981; 32(11): 843-845. CrossRef PubMed
- Stavchansky SA, Kostenbauder HB. Synthesis of carbon-14 labeled diphenylhydantoin and its major metabolite 5-(p-hydroxyphenyl)-5-phenylhydantoin (HPPH) in 70 minutes. J Label Compd. 1974; 10(3): 469-474. CrossRef
- Stavchansky SA, Tilbury RS, McDonald JM, Ting CT, Kostenbauder HB. In vivo distribution of carbon-11 phenytoin and its major metabolite, and their use in scintigraphic imaging. J Nucl Med. 1978; 19(8): 936-941. PubMed
- Hasegawa Y, Kanai Y, Hasegawa S, et al. Evaluation of brain and whole-body pharmacokinetics of 11C-labeled diphenylhydantoin in rats by means of planar positron imaging system. Ann Nucl Med. 2008; 22(4): 301-307. CrossRef PubMed
- Poupaert JH, Winand M, Dumont P. An efficient synthesis of 5-(phenyl-13C6)-5-phenylhydantoin. J Label Compd Radiopharm. 1981; 18(12): 1827-1829. CrossRef
- Casey DL, Gwilt PR, Digenis GA, Perrier DG. A convenient synthesis of deuterium labelled diphenylhydantoin. J Label Compd Radiopharm.1977; 13(4): 623-625. CrossRef
- Klumpp DA, Yeung KY, Surya Prakash GK, Olah GA. Superacid activated condensation of parabanic acid and derivatives with arenes. A new synthesis of phenytoin and 5,5-diarylhydantoins. SynLett. 1998; 8: 918-920. CrossRef
- Andresen BD, Biemann K. A synthesis of 5-phenyl-d5-phenyl-d5-hydantoin. J Label Compd Radiopharm.1978, 15(S1), 479-488. CrossRef
- Hoskins JA. The synthesis of deuterium labelled mono-hydroxy metabolites of phenytoin. J Label Compd Radiopharm. 1984; 21(7): 667-678. CrossRef
- Poupaert JH, Guiot P, Dumont P. Synthesis and biological evaluation of p,p′-dideuteriophenytoin J Label Compd Radiopharm. 1988; 25(1): 43-51. CrossRef
- Kepler JA. Preparation of tritium labeled diphenylhydantoin of high specific activity. J Label Compd. 1975; 11(4): 601-603. CrossRef
- Elvidge JA, Jones JR, Saljoughian M. Catalytic tritiation of drugs and analysis of the tritium distribution by 3H n.m.r. spectroscopy. J Pharm Pharmacol. 1979; 31(8): 508-511. CrossRef PubMed
- Poupaert JH, Claesen MH, Adline J, Dumont P. (+)-(R)- and (-)-(S)-5-(2-Deuteriophenyl)-5-phenylhydantoin: Synthesis, chiroptical properties, and absolute configuration. J Chem Res. 1979; 5: 174-175.
- Maguire JH, Butler TC, Dudley KH. Absolute configuration of (+)-5-(o-hydroxyphenyl)-5-phenylhydantoin, the major metabolite of 5,5-diphenylhydantoin in the dog. J Med Chem. 1978; 21(12): 1294-1297. CrossRef PubMed
- Coombe RG, Poulton DB. Preparation of 5,5-Bis(2-deuterophenyl)hydantoin. Aust J Chem. 1978; 31(2): 451-453. CrossRef
- Paxton JW, Rowell FJ, Ratcliffe JG. The evaluation of a radioimmunoassay for diphenylhydantoin using an iodinated tracer. Clin Chim Acta. 1977; 79(1): 81-92. CrossRef PubMed
- Rowell FJ, Paxton JW, Aitken SM, Ratcliffe JG. The effect of label affinity on the sensitivity and specificity of a hapten radioimmunoassay: a comparison of three [125I]diphenylhydantoin radioligands with the 14C-labelled drug. J Immunol Methods. 1979; 27(4): 363-371. CrossRef PubMed
- Yost Y, Polnaszek CF, Holtzman JL. Application of spin labeling to drug assays. III. [15N, 2H13]2,2,5,5-tetramethylpyrroline-1-oxyl-3-carboxylic acid coupled to phenytoin. J Label Compd Radiopharm. 1983; 20(6): 707-717. CrossRef
- Ames MM, Castagnoli Jr. N. The synthesis of 13C-enriched α-methyldopa. J Label Compd. 1974; 10(2): 195-205. Reference Source
- Howard HR, Evans M, Sarges R. Synthesis of 2H-, 3H- and 14C-labelled CP-45,634 (sorbinil). J Label Compd Radiopharm. 1991; 29(6): 703-708. CrossRef
- Miura Y, Hayashida K, Chishima S, Yoshikawa M, Takeyama S. Synthesis of carbon-14- and deuterium-labeled trimebutine and metabolites. J Label Compd Radiopharm. 1988; 25(10): 1061-1072. CrossRef
- Yu H, Niles JC, Wishnok JS, Tannenbaum SR. Spirodihydantoin is a minor product of 5-hydroxyisourate in urate oxidation. Org Lett. 2004; 6(19): 3417-3420. CrossRef PubMed
- Poje M, Paulus EF, Rocic B. Oxidation of uric acid. 1. Structural revision of uric acid glycols. J Org Chem. 1980; 45(1): 65-68. CrossRef
- Allen P, Hickey M, Kingston L, Mather A, Wilkinson DJ. Use of simple stable labelled intermediates to produce complex isotopically labelled internal standards. J Label Compd Radiopharm. 2007; 50(5-6): 590-592. CrossRef
- Palnitkar SS, Bin B, Jimenez LS, Morimoto H, Williams PG, Paul-Pletzer K, Parness J. [3H]Azidodantrolene: synthesis and use in identification of a putative skeletal muscle dantrolene binding site in sarcoplasmic reticulum. J Med Chem. 1999; 42(11): 1872-1880. CrossRef PubMed
- Takada Y, Ogawa M, Suzuki H, Fukumura T. Radiosynthesis of [2-(11)C-carbonyl]dantrolene using [(11)C]phosgene for PET. Appl Radiat Isot. 2010; 68(9): 1715-1720. PubMed
- Kumata K, Ogawa M, Takei M, et al. Radiosynthesis of [13N]dantrolene, a positron emission tomography probe for breast cancer resistant protein, using no-carrier-added [13N]ammonia. Bioorg Med Chem. 2012; 20(1): 305-310. CrossRef PubMed
- Bauman A, Piel M, Höhnemann S, et al. Synthesis, labelling and evaluation of hydantoin-substituted indole carboxylic acids as potential ligands for positron emission tomography imaging of the glycine binding site of the N-methyl-d-aspartate receptor. J Label Compd Radiopharm. 2011; 54(10): 645-646. CrossRef
- Delatour T, Gremaud E, Mottier P, Richoz J, Arce Vera F, Stadler RH. Preparation of stable isotope-labeled 2-nitrobenzaldehyde derivatives of four metabolites of nitrofuran antibiotics and their comprehensive characterization by UV, MS, and NMR techniques. J Agric Food Chem. 2003; 51(22): 6371-6379. CrossRef PubMed
- Hood HT, Skellern GG. The synthesis of 3-methyl-2-[4-14C]thiohydantoin a pharmacologically active metabolite of the antithyroid drug methimazole. J Label Compd Radiopharm. 1982; 19(6): 779-782. CrossRef
- Hays SJ. Synthesis of carbon-14 labeled CI-926 and CI-927, new antihypertensives. J Label Compd Radiopharm. 1987; 24(4): 351-360. CrossRef
- Ye Y, Muller JG, Luo W, Mayne CL, Shallop AJ, Jones RA, Burrows CJ. Formation of 13C-, 15N-, and 18O-labeled guanidinohydantoin from guanosine oxidation with singlet oxygen. Implications for structure and mechanism. J Am Chem Soc. 2003; 125(46): 13926-13927. PubMed
- Küpfer A, Bircher J. Stereoselectivity of differential routes of drug metabolism: the fate of the enantiomers of [14C]mephenytoin in the dog. J Pharmacol Exp Ther. 1979; 209(2): 190-195. PubMed
- Huguenin PN, Kelley JA. The synthesis of spiromustine-d8. A general approach to octadeuterated nitrogen mustards. J Label Compd Radiopharm. 1985; 22(8): 787-797. CrossRef
- Sharif NA, Williams GW, Davis TL. Pharmacology and autoradiography of human DP prostanoid receptors using [(3)H]-BWA868C, a DP receptor-selective antagonist radioligand. Br J Pharmacol. 2000; 131(6): 1025-1038. CrossRef PubMed
- Yu H, Wishnok JS, Tannenbaum SR. Synthesis of 3,7,8-15N3-N1-(β-D-erythro-pentofuranosyl)-5-guanidinohydantoin. J Label Compd Radiopharm. 2003; 46(14): 1269-1277. CrossRef
- Van Dort ME, Jewett DM, Jung Y-W, Sherman PS, Kuszpit KK. Synthesis of a carbon-11 labeled nonsteroidal antiandrogen as a potential radioligand for pet imaging of prostate cancer. J Label Compd Radiopharm. 2001; 44(S1): S330-S332. CrossRef
- Raja SN. Synthesis of [14C]ABT-770, matrix metalloproteinase inhibitor (MMPI), labelled in the phenoxy ring. J Label Compd Radiopharm. 2003; 46(9): 883-892. CrossRef
- Wheeler WJ, O’Bannon DD, Kennedy JH, Monn JA, Tharp-Taylor RW, Valli MJ, Kuo F. The synthesis of isotopically labeled (+)-2-aminobicyclo[3.1.0]hexane-2,6-carboxylic acid and its 2-oxa- and 2-thia-analogs. J Label Compd Radiopharm. 2005; 48(8): 605-620. CrossRef
- Tran SB, Maxwell BD, Chen S-Y, et al. Synthesis of lead LFA-1 antagonist [14C]spyrocyclic hydantoin. J Label Compd Radiopharm. 2009;52(6): 236-242. CrossRef
- Latli B, Byrne D, Nummy L, Krishnamurthy D, Senanayake CH. Synthesis of potent lymphocyte function-associated antigen-1 inhibitors labeled with carbon-14 and deuterium, part 1. J Label Compd Radiopharm. 2011; 54(12): 763-768. CrossRef
- Ono M, Hayashi S, Matsumura K, et al. Rhodanine and thiohydantoin derivatives for detecting tau pathology in Alzheimer’s brains. ACS Chem Neurosci. 2011; 2(5): 269-275. CrossRef
Article information
Corresponding author: Simon G. Patching.
Copyright: © 2017 Patching SG.
How to cite: Patching SG. Synthesis, NMR analysis and applications of isotope-labelled hydantoins. J. Diagn. Imaging Ther. 2017; 4(1): 3-26 (https://dx.doi.org/10.17229/jdit.2017-0225-026).
Article history: Received 06 January 2017; Accepted 25 January 2017; Published online 25 February 2017.
Archive link: JDIT-2017-0225-026
home »