REVIEW ARTICLE
Open Medscience, County Armagh, BT62 2LG, United Kingdom
Abstract
Squaryl molecular metaphors is the application of squaric acid building blocks towards creative functional group chemistry to produce lead compounds and imaging agents. This strategy is applied to rational drug design and to various imaging agents that would normally contain conventional functional group chemistry. These, include carboxylic acids, α-amino acids, peptide bonds and phosphonates. Moreover, the squaryl metaphor is a precursor to complex organic molecules involving functional group interchange (FGI) and rearrangements. This article discusses the application of these metaphors in the area of neurochemistry, especially NMDA receptors. An important area of drug design is the inhibition of angiotensin II enzyme by the use of losartan. This commercial drug contains a tetrazole moiety that can be modified using the squaryl group to give a semisquarate derivative. These concepts can extend to the semisquarate of ibuprofen. Moreover, a discussion on peptidomimetics regarding the substitution of the peptide bond for squaramide allows for a change in the biological properties due to modification at the peptide bond. Furthermore, the topic on how squaryl metaphors can be exploited primarily by the central 1,3-hydroxyamide sequence to the generation of a novel class of HIV protease inhibitors. Also, squaryl metaphors can be used to develop potential inhibitors of glutathione and novel anti-migraine drugs. The discussion continues on the design of anticancer drugs: in particular, a novel class of metalloproteases, including phosphonates for semisquaramides, leading to squaryl nucleosides. This review concludes with the application of squaryl metaphors in the design of positron emission tomography (PET) and magnetic resonance imaging (MRI) agents towards theranostics.
Keywords: squaryl metaphors; bioisosteres; rational drug design; positron emission tomography imaging; magnetic resonance imaging
In memory of Djuka Erakovic 1934-2013
Introduction
Langmuir first devised the term isosterism in 1919 and this regards the physicochemical properties of the constituents of organic molecules [1]. The concept of isosterism was developed further by Grimm, who applied the hypothesis of hydride displacement law to describe the ability of individual functional chemical groups to mimic other moieties [2,3].
Erlenmeyer rationalised these concepts and categorised isosteres into atoms, ions and molecules based on the valence level of electrons [4]. A further understanding by Friedman led to the term bioisosterism to include all atoms and molecules that fit the broadest definition of isosteres [5]. This approach was independent of whether the drug was an agonist or an antagonist towards biological activity. Moreover, Thornber applied the term bioisosterism to include subunits, groups or molecules that possess physicochemical properties of similar biological effects [6]. Finally, in 1991 Burger widened the definition of bioisosteres to include compounds or groups that possess similar molecular shapes and volumes independent of agonists or antagonists [7].
These bioisosteres would require approximately the same distribution of electrons and give a high probability of generating comparable physical properties such as hydrophobicity [8].
Currently, bioisosterism is one of the most useful tools to the medicinal chemist in rational drug design and in particular regarding the carboxylic acid bioisosteres [9]. The carboxylic acid functional group is part of the pharmacophore of many commercial drugs ranging from non-steroidal anti-inflammatory medicines (NSAIDs), statins, β-lactam antibiotics and anticoagulants [10]. The important role of the carboxylic acid group is its contribution towards biomolecular recognition at the receptor site, due to its ability to form strong hydrogen bonds [11].One lesson that can be apparent from drug discovery is that structures with the same bond connectivity/shape as the naturally occurring intermediates are not necessarily the most potent inhibitors [12]. A term commonly used to identify structures that can be used to replace others in bioactive molecules is bioisosterism [13]. It directly translates as the same shape as one present in a biological molecule. Here we may refer to these kinds of replacements as molecular metaphors (S. L. Kitson, Squarate Molecular Metaphors – A Strategy for Drug Design, personal communication, February 2001; S. L. Kitson, Molecular Metaphor Drugs – A New Approach to Rational Drug Design, personal communication, December 2001).
In the context of this review, molecular metaphors are described as the replacement of functionalities in a given substrate that can serve as molecular substitutes. These replacements may have similar structures but have different molecular recognition patterns. Therefore, the key to molecular recognition in biological systems, is the charge distribution of the substrate in space and not its molecular shape and volume [11]. Bioisosterism signifies that two molecules are similar and it is a term that would be better to fall into disuse regarding drug discovery [14–16].
A squaryl entity (Figure 1) is a molecular mimic that can undergo a functional group interchange (FGI) – the replacement of one functional group by another – to squaryl functionality [17]. A new compound is produced leading to a different or improved biological activity. The vast majority of pharmaceutical drugs are carbogenic compounds [18].
Furthermore, in vivo activity of these compounds depends on the solubility profile of plasma and tissue distribution to influence the associated physicochemical properties [19].
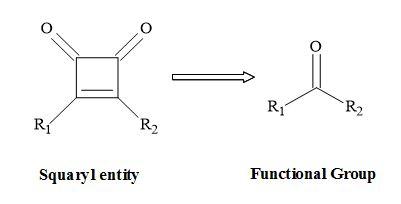
Figure 1. Squaryl molecular metaphors.
Conversely, the interaction of a drug with a receptor or an enzyme is dependent on the characteristics of the drug molecule. These factors include ionisation [20], electron distribution [21], polarity [22] and electronegativity [23]. This review discusses how to apply squaryl molecular metaphor technology to rational drug design and imaging agents. These types of molecular metaphors (Figure 2) relate to the function of squaric acid [24]. These include squarate, semisquarate, squaramic acid, squaramide [25] and squaric acid N-hydroxylamide [26]. Also, these metaphors can act as mimics for carboxylic acids [27], carboxylic esters [28], amino acids [29], peptides [30] and hydroxamic acid analogues [31].
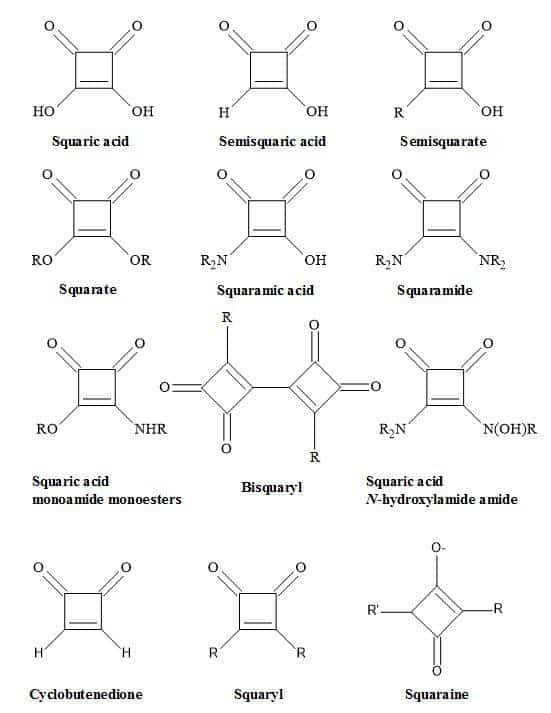
Figure 2. Nomenclature of squaryl entities.
These drug modifications produce a different biological activity profile [32]. Consequently, there may be a multitude of molecular modifications by the incorporation of squaryl entities leading to an increase in the drugs’ potency. For example, the synthesis of compounds bearing an aryl group can undergo substitution with the following groups: CH2SO2CH3; CH2SO3Et; CH2NHSO2CH3; CH2SO2NHCH3; CH2SO2NH2; CO2H; CH2CO2H and CH2CONH2. These moieties can substitute for a squaryl entity.
In general, carboxylic acid moieties can have detrimental effects on the volume of distribution [33]. Incidentally there are specific enzymes as evidenced by the short half-life for anti-inflammatory drugs. For example, ibuprofen and diclofenac have short half-lives of 2-3 hours compared to oxicams (e.g. piroxicam) half-lives of 20-60 hours [34].
The methodology behind squaryl molecular metaphors is to provide a novel technology for new leads in drug research. Many organic chemists have not taken up this challenge to date. Admittedly, it is hard to stay on top of a still rapidly developing synthetic methodology and move to systems of biological complexity at the same time. Some opinions signify that we have a complete array of methods available with which to probe and investigate living systems, for example the use of computational methods applied to rational drug design [35].
Molecular metaphors can have a broad range of applications, from synthetic organic chemistry to nanotechnology platforms [36] and including biomaterials [37]. Consequently, this review provides a valuable contribution to the interest of drug discovery and molecular imaging groups. Furthermore, this review will assist in the projection of new chemical entities leading to commercial drugs and imaging agents to address various disease states.
Classical metaphors
For a Pharmaceutical Company to succeed it must have a lucrative drug pipeline [38]. To generate new chemical entities (NCEs), the principles of molecular metaphors can be applied where possible to functional groups. The concept of bioisosteres does not apply to the subject of molecular metaphors, but only to classical metaphors [39]. However, these classical metaphors are functional groups that have similar spatial and electronic character. In many cases, replacement of a group with a bioisostere results in a new compound that retains the activity of the parent [40]. This approach is standard in medicinal chemistry since it allows the generation of marketable analogues of a known drug that has an intellectual property composition of matter [41]. These classical metaphors are shown below in Table 1.
Table 1. Classical metaphors.
Non-classical metaphors
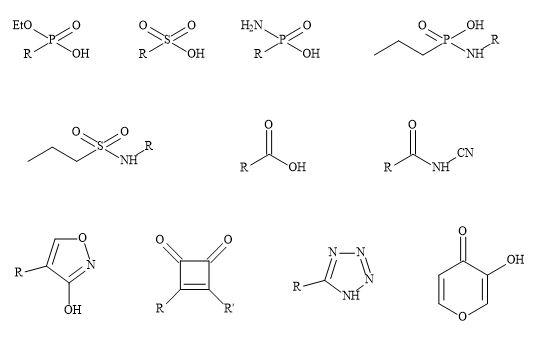
Non-classical metaphors do not have the same number of atoms but fulfil the steric and electronic rules of bioisosteres, however, do produce a similarity in biological activity [42]. This approach allows the medicinal chemist to make changes to the drugs’ parameters to augment potency and selectivity in order to produce a reasonable pharmacokinetics profile [43]. Multiple alterations may be necessary to counterbalance undesirable biological effects [44]. For example, modification of the functionality of the drug involved in binding may decrease the lipophilicity and its ability to penetrate the cell wall and diffuse across other membranes [45]. Subsequently, the drug molecule can undergo further substitution with additional lipophilic groups at sites distant from that are involved in receptor binding [46].
Consequently, modifications of this sort may change the overall molecular shape and result in other biological activity [47]. Carboxylic acid moieties are essential for biological activity; in particular for the substitution of the group with phosphonate, phosphonic and phosphinic acids, sulphonamides and tetrazoles (Figure 3) [48]. These subtle changes can have a major effect on the efficacy of the drug substrate [49]. For example, phosphonic and phosphinic acids have a higher acidity (pKa ~1-3) compared to carboxylic acids (e.g. aspirin pKa = 3.49) producing lower partition coefficients (log P) [50].
Figure 3. Non-classical metaphors.
Squaryl syllogism
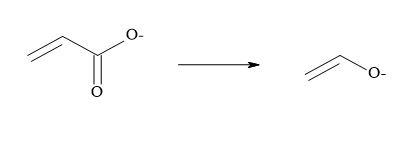
Metcalf et al. [51] and Holt et al. [52] have suggested that vinyl carboxylates can serve as equivalents for enolates and two other pharmaceutical groups have reported that semisquarates can be cognated to carboxylates [53,54]. Combining the above methodology that carboxylates are equivalent to enolates and semisquarates are equivalent to carboxylates then semisquarates can act as stable mimics for enolates.
Vinyl carboxylates can serve as equivalents for enolates:
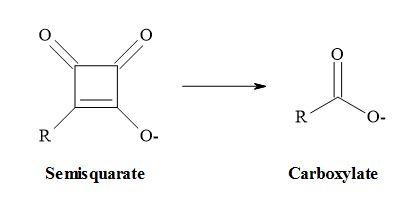
Semisquarates can be cognated to carboxylates:
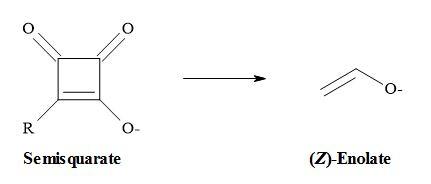
Combining these ideas that carboxylates are equivalent to enolates and semisquarates are equivalent to carboxylates. Therefore, semisquarates can act as a stable metaphor for enolates:
The second argument favours the semisquarate-for-enolate substitution in that like many 1,3-dicarbonyl compounds; semisquarates contain a stable enol within their structures. However, due to the constraint of its cyclic structure, the semisquarate can mimic only the (Z)-enolate [55].
Oxocarbons
The term oxocarbon was first coined in 1963 and designates compounds in which all of the carbon atoms connect to carbonyl or enolic oxygens, including the hydrated or deprotonated equivalents [56]. These cyclic oxocarbon anions are members of an aromatic series – (Hückel’s rule for monocyclic systems) – stabilised by the delocalization of electrons around the carbon ring [57]. The monocyclic oxocarbon family (Figure 4) include the following:
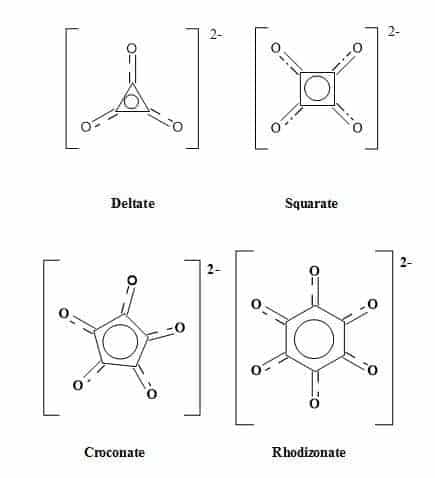
Figure 4. The oxocarbon family.
Numerous investigations have studied the structure and bonding of oxocarbons [58,59]. The monocyclic oxocarbon dianions (CnOn2-) and the neutral cyclic compounds (CnOn) comprise two prototype sub-groups within the oxocarbon family. The oxocarbons croconate (C5H52-) and rhodizonate (C6O62-) were discovered over 160 years ago. The lower analogues squarate (C4O42-) and deltate (C3O32-) were synthesised over the last few decades [60]. The aromaticity of (CnOn2-) (n = 3-6) decreases with increasing ring size. The deltate (C3O32-) is both σ and π aromatic, and squarate (C4O42-) is moderately aromatic while (C5H52-) and (C6H62-) are less aromatic [61].
Deltic acid
Deltic acid (dihydroxycyclopropenone) is a triangular oxocarbon first discovered in 1975 by Eggerding and West [62]. Deltic acid (H2C3O3) dissociates in water at 25 °C to give pKa1 = 2.57 and pKa2 = 6.03 resulting in a D3h symmetric deltate anion, (C3O3)2−. These pKa values indicate that deltic acid is a weaker acid than squaric acid [63]. One method to synthesise deltic acid is from squaric acid involving the formation of bis(trimethylsilyl)squarate using the reagent BSA [(N,O-bis(trimethylsilyl)acetamide)] (Figure 5).
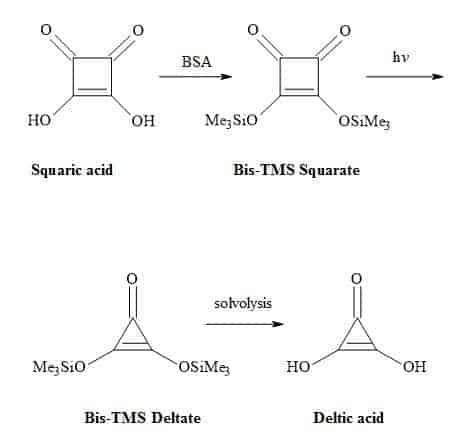
This intermediate is then subjected to photochemical decarbonylation to give bis(trimethylsilyl)deltate followed by solvolysis to generate the deltic acid [64].
Figure 5. Synthesis of deltic acid.
Squaric acid
Squaric acid (3,4-dihydroxy-3-cyclobutene-1,2-dione) also known as quadratic acid is a symmetrical D4h planar diprotic four-membered oxocarbon compound [51]. Although it is a small ring molecule, it possesses unique 2π-pseudo-aromaticity [65].
The acidity constants of squaric acid have been determined in solutions with low varying ionic strengths by Ireland and Walton to be pKa1 = 1.7 and pKa2 = 3.21 [66]. However, Tedesco and Walton reported pKa1 = 0.4 and pKa2 = 2.89 [67] and by MacDonald the acidity constants were pKa1 = 1.2 and pKa2 = 3.48 [68]. The work of Park et al. produced a value of pKa2 = 3 [69]. Alexandersson and Vannerberg [70] have reported the values of pKa1 = 0.96 and pKa2 = 3.19. This results in high acidity (pKa1 = 1.2-1.7, pKa2 = 3.2-3.5). The accepted acidity constants of squaric acid are pKa1 = 0.54 and pKa2 = 3.48 [71] and intrinsic properties of an organic acid [72] compared with sulphuric acid (pKa1 = -3.0 and pKa2 = 1.92) as shown in Figure 6.
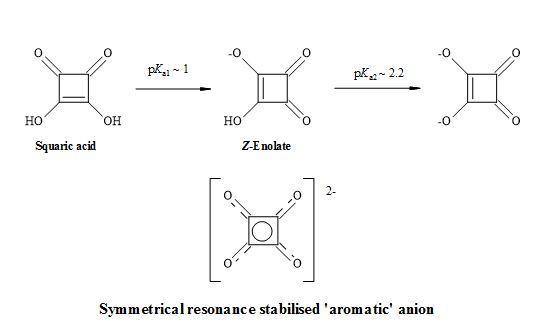
Figure 6. Resonance structures of squaric acid.
This inherent property of squaric acid makes it more anionic than other enols such as phenol (pKa ~10) or cyclopentane-1,3-diones (pKa ~6) at physiological pH. It contains a stable enol within the structure and due to the geometry of its cyclic structure, squaric acid can mimic only the (Z)-enolate form [73].
The (Z)-enolate and dianion of squaric acid is a valuable, versatile precursor to various ‘shapes’ in the synthesis of carbogenic compounds, due to the inherent ring strain of the cyclobutenedione structure [74]. The acidic hydroxy groups can undergo further transformations to provide esters [75], amines [76] and halide derivatives of squaric acid [77].
Various nucleophiles can give rise to 1,2- [78] and 1,4- Michael addition products at the carbonyl or olefinic carbon centres of the ring system [79]. Furthermore, the cyclobutenedione ring can be further transformed by thermolysis [80], photolysis [81] and the application of organocatalysts [82] to generate highly functionalized compounds.
Examples of these transformations are shown in Figure 7 and include (±)-septicine [83], perezone [84], lawsone [85], (Z)-multicolanic esters [86] and (E)-basidalin [87]. Also, squaric acid has been found to catalyse reactions involving the N-substituted of pyrroles [88].
It is evident from these examples that squaric acid is a useful C4-synthon building block for the construction of biologically interesting heterocycles. Another interesting transformation starting from alkyl squarate includes a 4π-6π electrocyclic ring opening and ring close to give echinochrome A [89].
Figure 7. Squaric acid is a key C4-synthon leading to highly functionalized compounds.
Synthesis of natural products
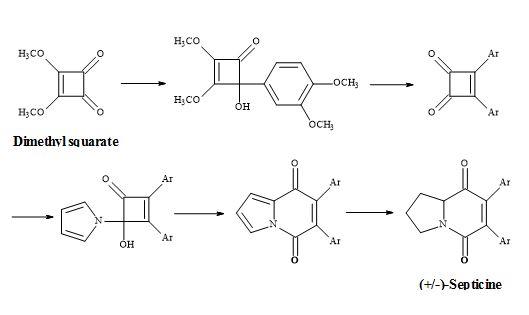
The total synthesis of (±)-septicine (Figure 8) was achieved in 8 steps in a yield of 27% starting from dimethyl squarate [83]. The synthesis involved the ring expansion of the 4-(1-pyrrolo)cyclobutenone to give indolizinedione system.
Figure 8. Synthesis of (±)-septicine.
The synthesis of this natural product, a toxic amine resulting from bacterial proteolysis [90] takes advantage of the asymmetry arising from the ring nitrogen atom [91].
Consequently, its synthesis is derived from a pyrrolidine moiety and with the correct regioselectivity to produce the (Z)-stilbene derivative. This approach benefits from the absence of regiochemical problems in the joining of these building blocks.
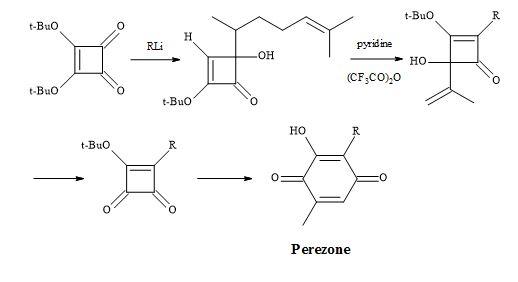
Furthermore, a route has been developed from dimethyl squarate via 3,4-bis(3,4-dimethoxyphenyl)-1,2-cyclobutenedione. Since the two carbonyl groups in the diketone are the same, the reaction with N-lithiopyrrole yields one product. Other natural product syntheses take advantage of the symmetrical properties of squaryl derivatives and skeletal changes via electrocyclic reactions and include perezone [84] and lawsone [85]. The syntheses of (E)-basidalin [86] and (Z)-multicolanic esters [87] demonstrated a new method to access substituted furanones (Figures 9–12).
The above compound shapes are prepared from squaric acid on a solid support involving palladium-mediated coupling reactions to produce multiple core structure libraries. The halogenated aryl ether on Wang resin are generated through a Mitsunobu reaction and subsequently coupled to tributyltin isopropyl squarate under Stille conditions. Another route involves 1,2-addition of nucleophiles to the squarate followed by acid-mediated rearrangement. In both cases, the product is cleaved from the resin under trifluoroactic acid conditions [74].
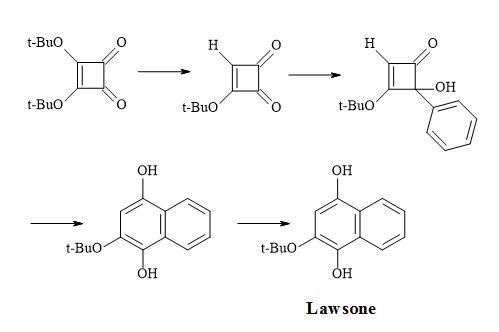
Figure 9. Synthesis of perezone.
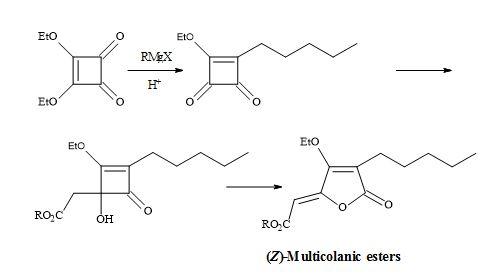
Figure 10. Synthesis of lawsone.
Figure 11. Synthesis of (Z)-multicolanic esters.
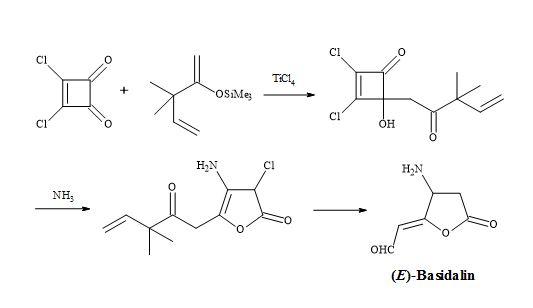
Figure 12. Synthesis of (E)-basidalin.
Moniliformin
In 1970, a strain of the mould Fusarium moniliforme was isolated from corn seed damaged by southern leaf blight [92]. The mould was found to produce water-soluble mycotoxin and given the trivial name moniliformin (Figure 13) [93]. This compound has growth-regulating and phytotoxic effects on mammals [94]. Moniliformin (pKa = 0.88) from the fungal plant pathogen Gibberella fujikuroi was found by X-ray analysis to be the potassium salt of the semisquaric acid [95]. Furthermore, semisquaric acid is an inhibitor of pyruvate dehydrogenase and transketolase compared to squaric acid which acts as an inhibitor of glyoxylase I. The original toxin is the corresponding sodium salt. The semisquaric acid molecule contains a vinylogous acid function linked to a carbonyl group in a four-membered ring system [96].
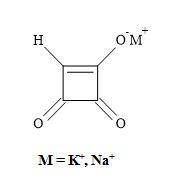
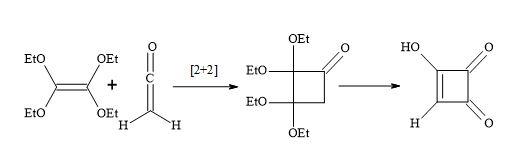
The [2+2]-cycloaddition of gaseous ketene to tetraethoxyethylene yields crystalline, stable cyclobutanone (Figure 14). Surprisingly, the hydrolysis of the cyclobutanone was not a straightforward reaction. Under varied experimental conditions, large amounts of ring-opened products were formed. Finally, the hydrolysis with 18% hydrochloric acid, in the presence of dioxane at 60°C, afforded semisquaric acid of 90% yield [96].
Figure 14. Synthesis of semisquaric acid
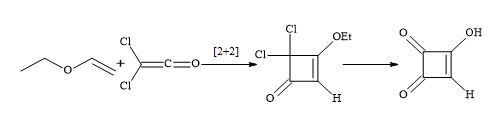
Spinger et al. accomplished a short semisquaric acid synthesis via a haloketene [97]. The [2+2]-cycloaddition of ethylvinyl ether with dichloroketene formed in situ from dichloroacetyl chloride and trimethylamine, afforded the cyclobutenone derivative that could then be hydrolysed by aqueous hydrochloric acid to semisquaric acid (Figure 15). In both cases, the semisquaric acid is readily converted into the salt form to afford moniliformin.
Figure 15. Synthesis of semisquaric acid
Squaric acid family
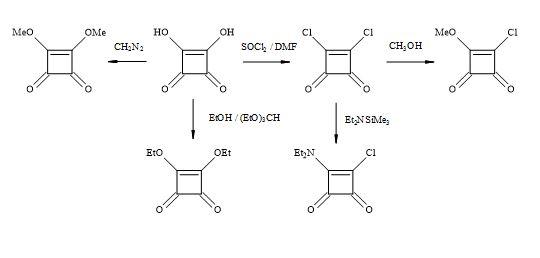
Squaric acid is a four-membered oxocarbon compound that has potentially useful multi-functionality and intrinsic ring strain towards the synthesis of complex compounds. Squaric acid has been the focus of several research groups including West, Kinney, Liebeskind, Moore and Paquett [98]. Based on the cyclobutenedione structure, acidic hydroxyl groups are replaced by superior leaving groups and then various nucleophiles can react at the carbonyl or olefinic carbon centres.
The squaric acid family of derivatives consists of dichloride, methyl ester chloride, diethylamide chloride, alkyl esters (Figure 16). These squaryl derivatives are versatile building blocks in the synthesis of new chemical entities towards the treatment of chronic inflammatory diseases such as rheumatoid arthritis, multiple sclerosis and asthma [99].
Figure 16. Squaric acid family.
Synthesis of squaric acid
Squaric acid was first synthesised by Cohen et al. in 1959 from the aqueous hydrolysis of 1,3,3-triethoxy-2-chloro-4,4-difluorocyclobutene. Furthermore, by the aqueous and acid hydrolysis of 1,2-diethoxy-3,3,4,4-tetrafluorocyclobutene [100]. Interestingly, the carbon-14 radiosynthesis of squaric acid reported by Heys and Chew [101] was modelled on the synthetic route reported by Bellus [102]. This methodology involved a [2+2] cycloaddition of tetra-alkoxyethylenes with oxyketenes.
These oxyketenes generated in situ by triethylamine-promoted dehydrohalogenation of the corresponding acyl chlorides. The cyclobutanone intermediate formed undergoes rapid enolization followed by esterification with another equivalent of acid chloride to give the key compound which is the cyclobutenol ester. Displacement-induced fragmentation of the ester, promoted by SiO2/triethylamine or basic Al2O3, yields smoothly the mono-orthoester of squaric acid. The acid hydrolysis of the cyclobutenol ester and the orthoester afford [U-14C]squaric acid in high yield (Figure 17).
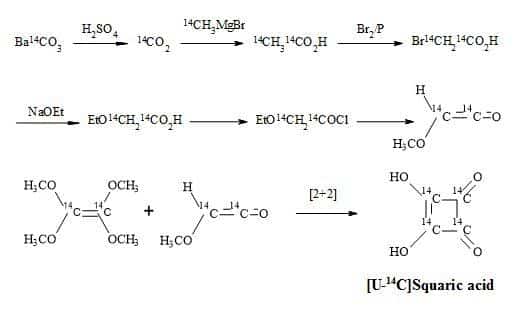
Figure 17. Synthesis of [U-14C]squaric acid.
The lowest member of this series of oxocarbons, methylmoniliformin (3-hydroxy-4-metbylcyclobut-3-ene-1,2-dione) was prepared first by Chickos [103] via the addition of methylmagnesium bromide to the substrate, diethyl squarate (Figure 18).
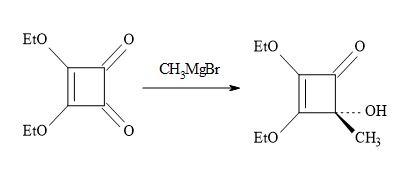
Figure 18. Synthesis of methylmoniliformin.
Previous studies on the reaction of Grignard reagents with dimethyl squarate resulted in the isolation of mono- or dialkyl- cyclobutenedione derivatives according to a 1,4- addition process (Figure 19). Kraus et al. [104] have discovered that lithium reagents react with dimethyl squarate at the carbonyl centre via a 1,2-addition process leading to 2-hydroxy-3,4-dimethoxy-3-cyclobutenones.
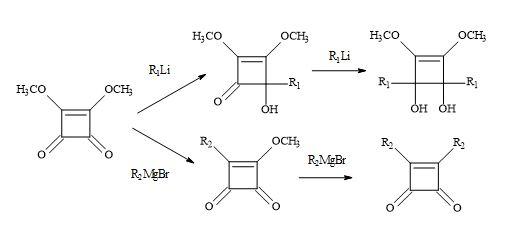
Figure 19. Michael addition of Grignard reagents and lithium reagents to dimethyl squarate.
Based on several literature precedents, it appears that lithium reagents attack dimethyl squarate exclusively at the ketone function. Furthermore, Grignard reagents favour conjugated addition [98]. When there is a competition between the direct attack on the ketone function (1,2-addition) and conjugate addition of a α-β-unsaturated ketone substrate, the less stable carbanion favours 1,2-addition. Consequently, the more resonance stabilised carbanion favours conjugation addition. Dehmlow and Schell [105] reacted several dialkyl squarates, for example, di-ethyl, di-n-propyl, di-isopropyl, di-n-butyl, di-tert-butyl and di-benzyl with various Grignard reagents. On acid hydrolysis, the corresponding 3-alkyl-4-alkyl-3-cyclobutene-1,3-diones were isolated in yields ranging from 2% to 65%. The lower yields were due to the formation of 1,2- and 1,4-addition products and by over addition of the Grignard reagents to the dialkyl squarate (Figure 20).
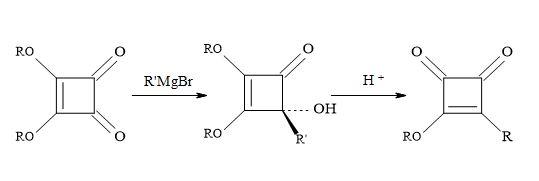
Figure 20. Addition of Grignard reagents on di-propyl squarate.
Liebeskind and co-workers chose to work with di-isopropyl squarate. This material prepared in 86% yield by refluxing squaric acid in excess toluene/propan-2-ol [106]. The di-isopropyl squarate reacts with the following Grignard reagents: RMgX/yield: EtMgX (43%), iso-PrMgX (65%) and tert-BuMgX (14%). Then on subsequent hydrolysis to give moderate yields of 3-(1-methylethoxy)-4-alkyl-3-cyclobutene-l,2-diones. This chemistry seldom proceeded cleanly and required the isolation of product by careful chromatography. Other procedures used commercially available di-isopropyl squarate to react with the di-isopropyl magnesium chloride (R3MgCI) and iso-butylmagnesium bromide (R4MgBr) to afford the corresponding 4-hydroxy-4-substituted-2,3-bis(l-methylethoxy)-2-cyclobuten-1-ones [55,107].
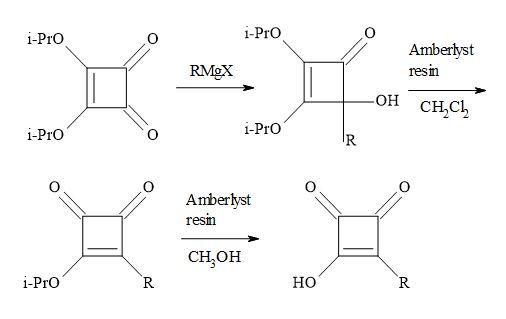
Figure 21. Resin-H+ work-up.
These were not isolated and in turn converted into the 3-(1-methylethoxy)-4-substituted-cyclobut-3-ene-1,2-diones, simply by stirring in dichloromethane – in the presence of dry amberlyst-15 ion exchange resin – in a two-phase system at ambient temperature. The conversion of this semisquaric acid derivative, was facilitated using wet amberlyst-15 ion exchange resin, simply by stirring in methanol at ambient temperature (Figure 21).
Squaryl molecular metaphors
This section outlines the methodology behind classical and non-classical metaphors. The broadest definition of classical metaphors (bioisosteres) consists of groups or molecules having similar chemical and physical resemblances to produce comparable biological properties [32].
A trend in this area is the increasing occurrence of non-classical metaphors that do not have the same number of atoms and produce a parameter that is vital to the receptor-drug interaction. Therefore, similar effects in two functional groups do not imply atom upon atom overlap [42].
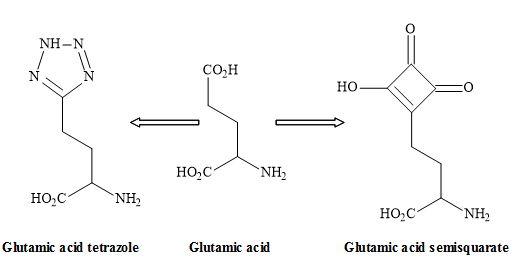
Figure 22. Tetrazole – semisquarate interchange.
The tetrazole group shown in Figure 22 is an excellent non-classical metaphor for the carboxylic acid moiety of glutamic acid [108]. Other carboxylic acid metaphors include the phosphinic or phosphonic acid [109] and the sulphate ester functionality (Figure 23) [40].
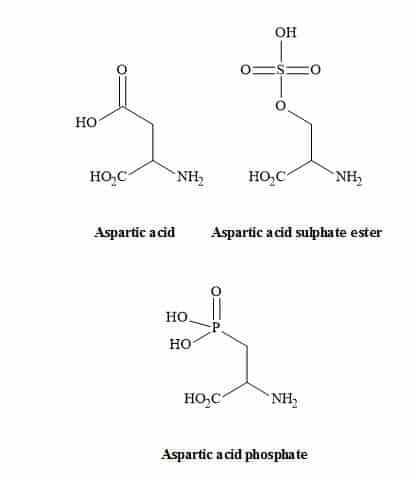
Figure 23. Carboxylic acid metaphors.
Figure 24 outlines the rational thinking behind the principles of squaryl molecular metaphors that describe the transformations of carboxylic acids, amino acids and peptides to their corresponding squaryl derivatives.
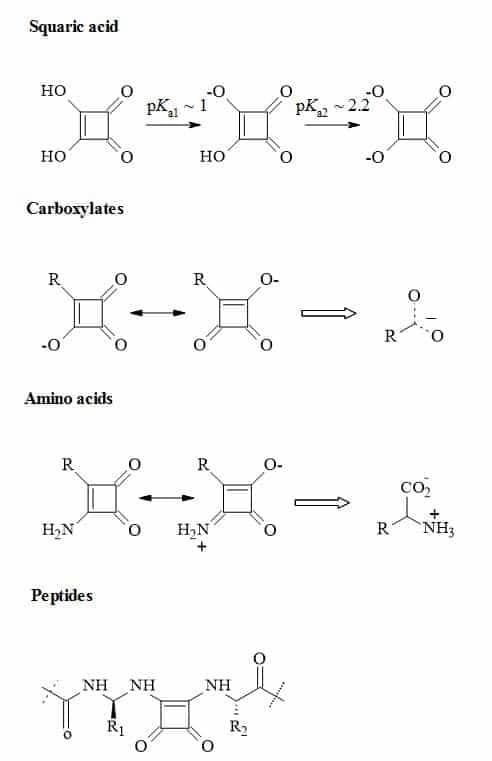
Figure 24. Metaphors of carboxylic acids, amino acids and peptides to their corresponding squaryl derivatives.
The semisquarate (Figure 25) can offer en routes to novel chemical entities that can provide the medicinal chemist new types of functional groups to generate lead compounds. The resultant substituted semisquarates can mimic the unstable equivalent and therefore obtain different electronic properties. These changes in property may result in new interactions with the active site in proteins and other biomolecules [110].
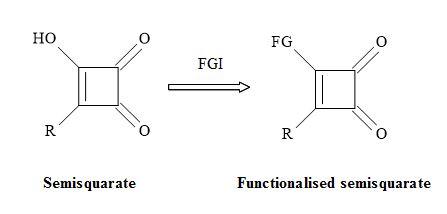
Figure 25. Functionalisation of semisquarate.
The semisquarates of cyanides and amino derivatives should be relatively stable in the ring form. These masked compounds in the squaryl entity would benefit further chemical transformations especially allowing for conjugation of biomolecules. These may include monoclonal antibodies, proteins and novel anti-nicotine vaccines [111].
Table 2 demonstrates the potential usage of squaryl molecular metaphors showing a variety of transformations to give carbonyl functionality. These squaryl molecular metaphors can substitute the peptide bond and peripheral functional groups sites on the peptidic drug [14,15]. Table 2 provides various examples of the synthetic equivalent such as the squaryl carboxylic acid (R1 = R, R2 = OH) to the corresponding carboxylic acid. Some of these resultant compounds would normally be unstable. However, the squaryl derivative may well be stable with notably higher acidic properties, in the case of carboxylic acids. Interestingly, these principles can be extended to include the replacement of N-cyanoguanidine moiety in a drug lead for squaramide [112,113].
Table 2. Examples of functional group equivalents.
Other squaryl metaphors (thioxocarbons) can be used to mimic thiolketones shown in Figure 26 [114].
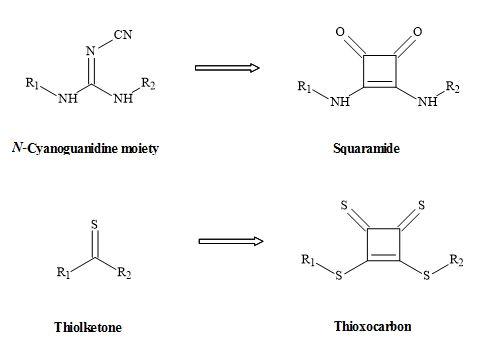
Figure 26. Squaryl metaphors of thiolketones and N-cyanoguanidine.
Pyrrolyl squaraines
The pyrrolyl squaraine chemical structure is present in many dyes and polymers and first appeared in literature in 1965 by Treibs and Jacob [115]. Since that time these pyrrolyl squaraines have generated interesting compounds, having both optical and electrical conduction properties in addition to magnetic properties [116]. The synthesis of pyrrolyl squaraines occurs between the condensation of a pyrrole (or linked bispyrrole) and squaric acid (Figure 27).
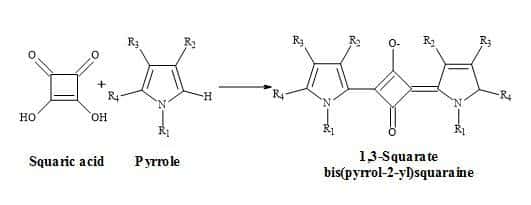
Figure 27. Synthesis of bisquaryls.
Squaryl amino acids
A squaryl mimic for the α-amino acid group might provide greater bio-availability or brain penetration for these agents relating to the amino acid class of Excitatory Amino Acids (EAA) antagonists [117]. At physiological pH, the α-amino carboxylic acid group is present as an ammonium carboxylate [118]. The squaryl chosen to mimic amino acid functionality is squaramide (3,4-diamino-3-cyclobutene-1,2-dione) [10].
The squaramide contains a dipole, possssing a partial negative charge on the carbonyls and a partial positive charge on the nitrogen. The dipole is a contribution from the canonical forms [57].
Though the 3,4-diamino-3-cyclobutene-1,2-dione group resembles the ammonium carboxylate functionality electronically; it should be emphasized that it lacks other properties of α-amino carboxylic acid (Figure 28). For instance, the group is not basic or acidic at physiological pH and the amino group is not nucleophilic. A potential advantage of this mimic over α-amino acid is its lack of chirality, making these derivatives more readily accessible.
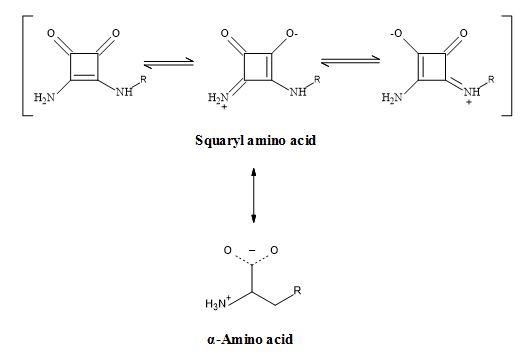
Figure 28. Squaramide – amino acid synergy.
Hence, the group 3,4-diamino-3-cyclobutene-1,2-dione contains a dipole making it an electronic metaphor of the ammonium carboxylate. The Figure 29 below outlines the squarate metaphors methodology for potential proteinogenic α-amino acids.
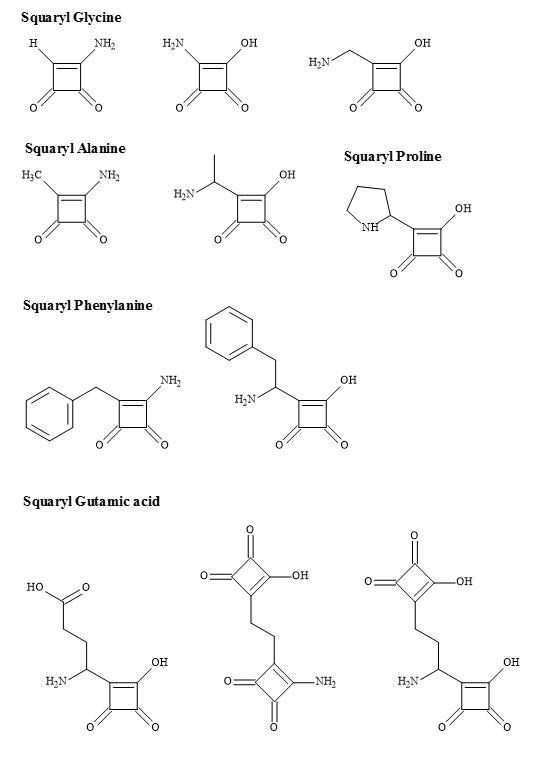
Figure 29. Squaryl amino acid families.
The β-amino acids – although of less importance than the parent α-amino acids – are crucial structural features of numerous biologically active natural products, as well as building blocks for β-lactam antibiotics [119].
Furthermore, the squaryl amino acids are accessible from the initial addition reaction of a dianion enolate, generated from N-Boc α-amino acid tert-butyl ester, to diisopropyl squarate, followed by the subsequent decarboxylation of the resulting carboxylic acid moiety [29] shown in Figure 30.
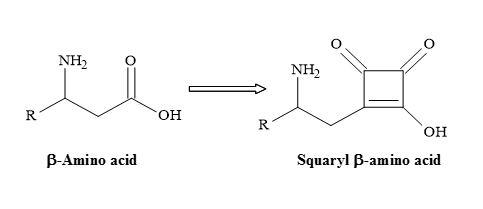
Figure 30. Semisquarate β-amino acids.
A new set of amino acids could in principle result from the substitution of the carboxylic moiety for a semisquarate entity (Figure 31).
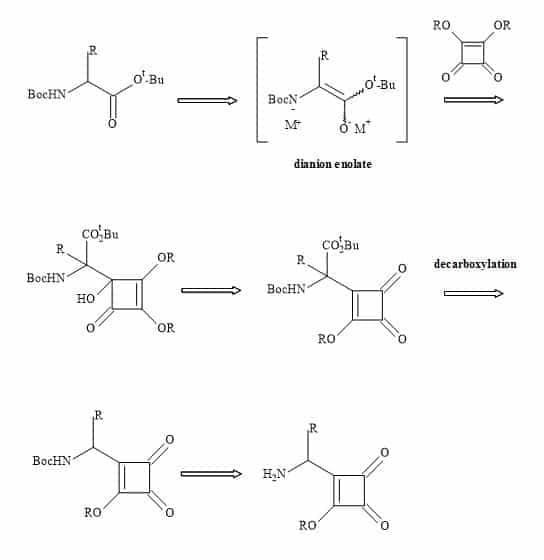
Figure 31. Synthesis of squaryl amino acids.
The squaryl amino acids in Figure 32 are metaphors of the natural proteinogenic amino acids synthesised by a radical reaction between 2-stannylcyclobutenedione and the carbanion addition to di-isopropyl squarate.
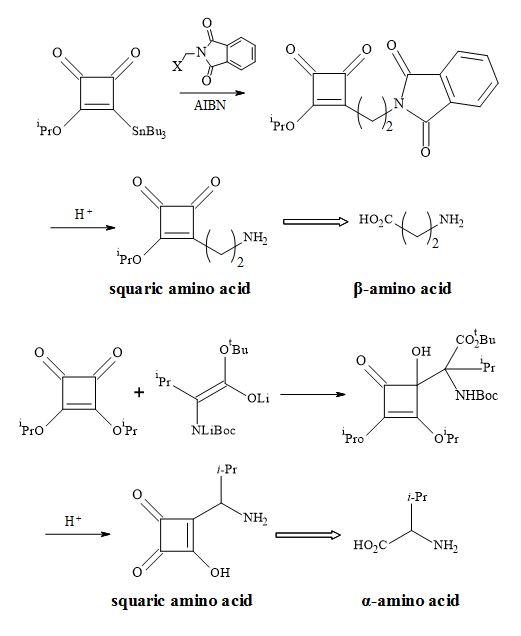
Figure 32. Synthesis of squaryl amino acid metaphors.
Neurochemistry
The N-methyl-D-aspartate (NMDA) receptor is a site for the neurotransmitter glutamate, which is the most important excitatory transmitter in the brain [120]. NMDA also acts as a ligand-gated ionic channel [121] and is the famous agonist of the NMDA receptor [122]. The receptor is very complex and the ligand-gated channel seems too implicated in the toxic effects of excessive glutamate [123]. These cellular functions include (Figure 33) [124]
- the postsynaptic receptor for L-glutamate, (the major excitatory neurotransmitter in the CNS, that can be activated by the drug NMDA;
- both voltage sensitive and ligand-gated channel for calcium;
- synaptic plasticity and memory formation;
- excessive Ca2+ enters the postsynaptic neuron via NMDA receptors, it exerts toxic effects on that cell by activating second messenger systems that result in the apoptosis of the cells.
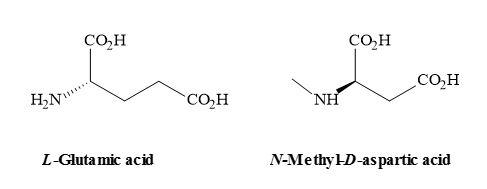
Figure 33. Chemical structures of L-glutamic acid and NMDA.
The NMDA receptor plays a role in neurodegenerative disease states. These include the following: excitotoxicity [125] – over stimulation of the central glutamate receptors, resulting in a slowly progressive neural death both in vitro and in vivo. This excitotoxicity is believed to contribute to neurodegenerative disease states [126].
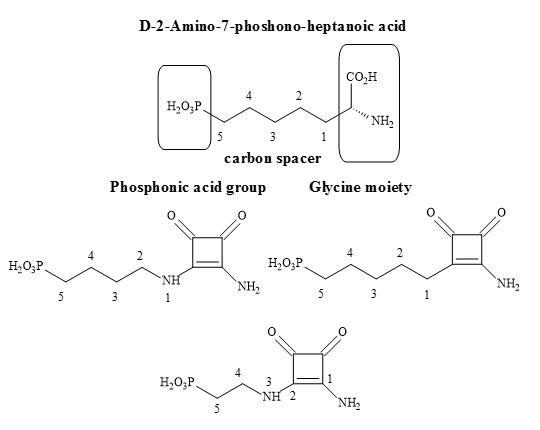
Figure 34. Potential squaramide NMDA antagonists.
These include both acute and progressive neurological disorders, such as seizures. The blockade of glutamate receptors (including the NMDA receptor), can decrease excitability and reduce seizure activity [127]. Investigations into NMDA receptor blockade is a possible way to reduce the damage that occurs from episodes of seizure. Also, it has been shown that NMDA receptor antagonists can induce schizophrenia-like psychosis [128].
All NMDA receptors possess several binding sites of pharmacological interest including those for glutamate, glycine, polyamines and open channel blockers [129]. A novel squaryl molecular metaphor of the amino acid, 3,4-diamino-3-cyclobutene-1,2-dione, has been incorporated into a series of NMDA antagonists [130]. These achiral compounds have demonstrated an affinity for the NMDA receptor, for example perzinfotel (EAA-090). Perzinfotel acts as a potent NMDA antagonist. It has neuroprotective properties and has been investigated for the treatment of stroke, but lacks analgesic effects. Nevertheless, it shows a reasonable safety profile compared to older drugs, and further development is required to produce a more favourable pharmacokinetic profile [131].
Outlined below is the synthetic methodology to the target squaryl compounds (Figure 34). Diethyl squarate can undergo a 1,4-Michael addition to the enolic double bond and then subsequent ejection of a mole of ethanol with aminoalkyl phosphonate esters (Figure 35). These phosphonate esters are readily prepared from the corresponding trivalent phosphoric acid esters [P(OR)3] using Arbuzov conditions. Ammonolysis of the phosphonate-squarate adduct, followed by selective hydrolysis of the phosphonate ester produced the phosphonic acid squaramide [96].
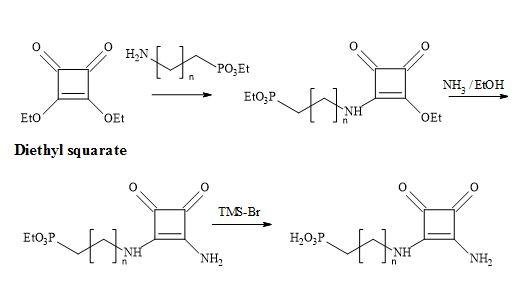
Figure 35. Synthesis of phosphonic acid squaramide.
The technology developed by Liebeskind [132] involves the treatment of 3,4-di-ethoxycyclobutenediones with the silylstannane, [n-Bu3SnSiMe3] under catalytic conditions [tetra-n-butylammonium cyanide] furnished 3-ethoxy-4-(tri-n-butylstannyl)cyclobutenedione in 65% yield (Figure 36). This compound is cross-coupled with organic iodides attached to sp2 and sp-hybridized carbon atoms, under the influence of co-catalytic palladium/Cu species, to provide 3-ethoxy-4- substituted-cyclobutenediones in excellent yields. Highly substituted and functionalised cyclobutenediones can be prepared under mild and neutral reaction conditions to form carbon-carbon bonds (Stille coupling) [133].
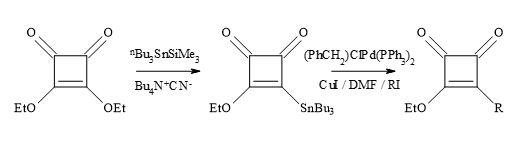
Figure 36. Liebeskind’s technology.
A novel free radical approach involves using mono- and di-alkyl-substituted cyclobutenediones has received much attention for its importance as synthetic precursors [96]. Recent procedures to prepare mono-substituted derivatives involve organolithium or Grignard addition to 3,4- dialkylcyclobutene-1,2-diones, and then either hydrochloric acid rearrangement to afford 3-alkoxy-4-alkylcyclobutene-1,2-diones [96].
An alternative to the synthesis of substituted cyclobutenediones, is a free radical approach developed by the research group of Kinney [130]. This strategy is used in the presence of the diethoxyphosphinyl group required for the synthesis of potential NMDA antagonists, as well as other functionalities [132]. Liebeskind’s reagent (3-ethoxy-4(tri-n-butylstannyl)cyclobutenedione) is useful in palladium-catalysed Stille cross-coupling with various alkyl and aryl iodides.
The alkyl radicals formed in the presence of a radical initiator, azobiscyclohexylnitrile (AIBN) on alkyl halides (Figure 37). The radical species first react with Liebeskind’s reagent, followed by the elimination of trialkyltin radical, to afford 3-alkoxy-4-alkylcyclobutene-1,2-diones in reasonable yields. This novel free radical procedure provides an additional method for incorporation of functionalized alkyl groups into the cyclobutenedione moiety [132].
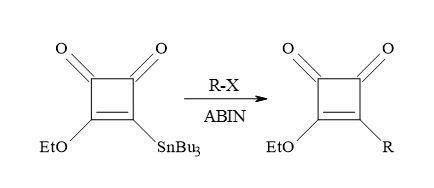
Figure 37. Synthesis of 3-alkoxy-4-alkylcyclobutene-1,2-diones.
The overall synthesis of the NMDA antagonist (Figure 33) involves the reaction between diethyl-(2-aminoethyl)-phosphonate and 3,4-diethoxy-3-cyclobutene-l,2-dione (diethyl squarate) in ethanol, to afford the phosphonate ester of monoethoxy squarate. This substrate, when treated with ethanolic ammonia, gave the 3,4-diamino-3-cyclobutene-1,2-dione moiety of the phosphonate ester. Deprotection of the phosphonate ester by refluxing in 1,2-dichloroethane included a large excess of bromotrimethylsilane, to afford the phosphonic acid squaramide [132].
Losartan semisquarate
The methodology of molecular metaphors can be a useful tool to the medicinal chemist in rational drug design. In particular, the carboxylic acid moiety has been of interest in biomolecular recognition. This concept can play a major role in the discovery of new non-peptidic angiotensin II antagonists [134]. Systematic evaluation of molecular metaphors for the replacement of the carboxylic acid moiety in Losartan, led to substitution with tetrazole, a more favourable anti-hypertensive profile, in comparison to its carboxylic acid congener [135].
Although an array of carboxylic acid metaphors exist and include tetrazole and other acidic heterocycles, hydroxamic acid and trifluorosulphonamide, there is moderate bioisosteric literature reported for the semisquarate derivatives. Other carboxylic acid metaphors include boronic acids, mercaptoazoles and sulfonimidamides.
In summary, it has been demonstrated that carboxylic acid molecular metaphors of 3-hydroxy-3-cyclobutene-1,2-dione, can give rise to new lead compounds, in the area of antihypertensive drugs [96]. These squaryl derivatives – coupled with the Liebeskind’s technology – make this an attractive area in the field of drug research.
The reaction involves Liebeskind’s reagent with alkyl or aryl halides under Pd/Cu catalysed cross-coupling Stille conditions. The treatment of 3,4-di-ethoxysquarate with n-Bu3SnSiMe3/catalytic cyanide, afforded Liebeskind’s reagent in 65% yield (Figure 38). This compound cross-coupled with organic iodides – attached to sp2– and sp-hybridized carbon atoms – to provide 3-ethoxy-4-substituted-cyclobutenediones in a reasonable chemical yield [136].
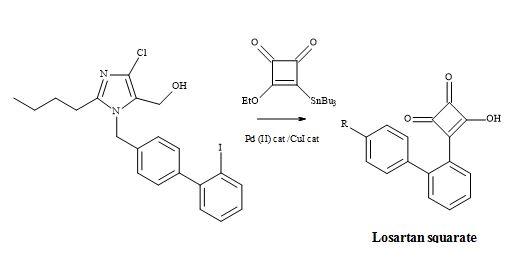
Figure 38. Synthesis of losartan semisquarate.
Hence, highly substituted and functionalized cyclobutenediones are prepared under mild reaction conditions, to generate carbon-carbon bonds. Subsequent acid hydrolysis on the monoethoxy group of the squarate produced the semisquarate and acts as a non-classical metaphor of the carboxylic acid group.
Ibuprofen semisquarate
NSAID ibuprofen has found widespread use since it showed improved potency and tolerance over aspirin, which had for decades been the mainstay drug for similar indications [137]. A vast demand for ibuprofen, combined with the relatively high dosage can amount to grammes per day. The substitution of the carboxylic acid moiety in ibuprofen for a molecular entity such as semisquarate (Figure 39) could potentially lead to more potent drugs and therefore a new family of NSAID lead compounds.
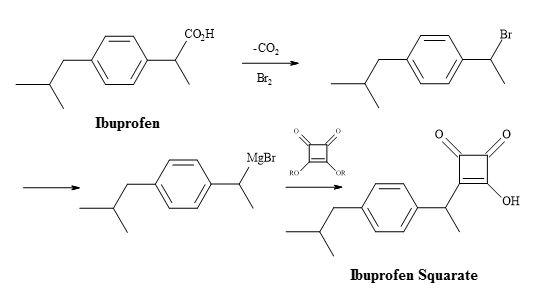
Figure 39. Synthesis of ibuprofen semisquarate.
Ibuprofen can undergo the decarboxylation reaction to give a secondary halide substrate. The conversion of this substrate – to the corresponding organometallic compound – can facilitate the addition to the dialkyl squarate. The resultant adduct is susceptible to acid hydrolysis of the ester, leading to the semisquarate of ibuprofen.
Suosan semisquarate
The suosan family of high-potency sweeteners was discovered by Muller and Petersen [138]. These sweeteners contain the N-aryl-N‘-(carboxymethyl)- or -(2-carboxyethyl)urea moieties and are both carboxylic acid mimics (Figure 40). The major disadvantage of the suosan sweetener family is their low water solubility, particularly at the pH used in carbonated beverages. A potential carboxylic acid metaphor is the use of a semisquarate derivative [139]. Incorporation of this highly acidic polar group, may well assist in the solubility of these new squarate sweeteners, in carbonated food preparations [140
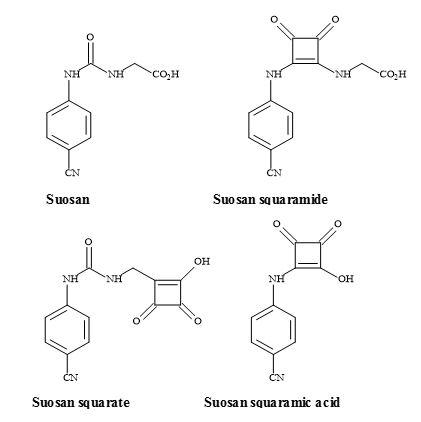
].
Figure 40. Suosan, semisquarate and semisquaramide.
Lactivicin squaramic acid
The antibiotic lactivicin (Figure 41) is not a typical β-lactam but can bind to penicillin proteins [141]. This binding indicates a similar mode-of-action to the β-lactam antibiotics family. The cyclic amide of lactivicin, is susceptible to activation by the electronegative oxygen, bonded directly to the nitrogen and the accompanying lactone moiety. The cyclic amide and the lactone moieties are capable of being substituted for squaryl entities. This modification in principle can activate the lactam amide, by the delocalization of the nitrogen lone pair. The squaryl moiety can also serve – not only as an amide-activating functionality – but also as a source of the required anionic centre.
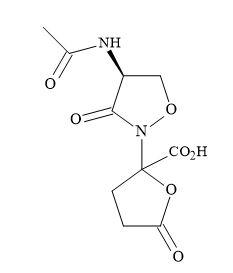
Figure 41. Lactivicin.
The starting material phenoxyacetyl-(L)-cycloserine, in the lactivicin squaramic acid synthesis, was prepared by Stammer et al. by the treatment of phenoxyacetyl-(L)-O-aminoserine hydrochloride with aqueous hydrochloric acid [142].
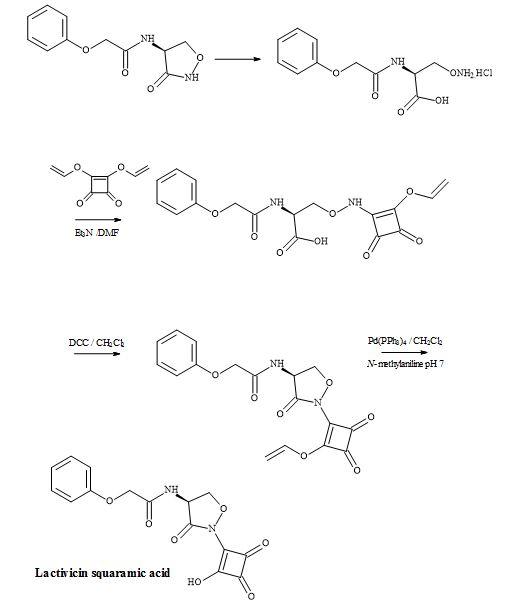
Figure 42. Synthesis of lactivicin squaramic acid.
This material was then treated with diallylsquarate, in the presence of triethylamine, to give O– (allyloxydioxocyclobutenyl)aminoserine in an overall yield of 37%. Acylation using the activating agent DCC proceeded smoothly to the acid sensitive cycloserine derivative. The allyl group was catalytically cleaved by palladium(0) in the presence of N-methylaniline to afford the target compound, lactivicin squaramic acid (Figure 42).
Vasotec semisquarate and squaramide
Angiotensin-converting enzyme (ACE) inhibitors are a family of peptides, of major significance, for controlling hypertension and congestive heart failure [143]. The majority of them possess a common structural element, an N-substituted ethyl-(S)-2-amino-4-phenylbutyrate moiety [144]. The chemical structure of some ACE inhibitors including quinapril, trandolapril and moexipril are shown in Figure 43.
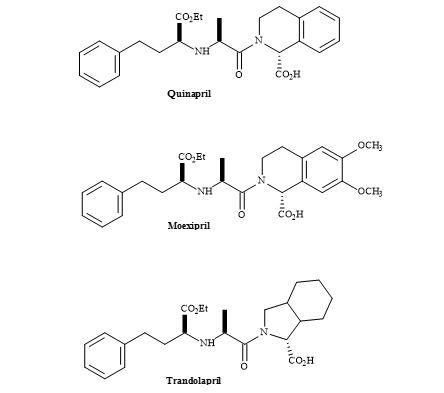
Figure 43. ACE inhibitors.
The syntheses of this group of ACE inhibitors have been reported via reductive amination of 2-oxo-4-phenylbutyrate, conjugate addition of amines to ethyl-4-oxo-4-phenylcrotonate followed by reduction, SN2 displacement of (R)-2-hydroxybutyric acid-derived triflate intermediates by amines and via asymmetric alkylations [145].
The ACE inhibitor vasotec (enalapril) was originally synthesized by Merck Sharp and Dohme for the treatment of hypertension. Vasotec contains a dipeptide alanyl proline residue and may be modified using squaryl molecular metaphors [146].
The methodology applied to lactivicin squaramic acid synthesis can be used to replace the proline moiety with a squaramide group. Alternately, the carboxylic acid functionality on proline can be replaced with a squarate group. These subtle changes may lead to a better drug profile than vasotec. This lead discovery demonstrates that squaryl entities are a powerful tool in the intellectual property of pharmaceuticals (Figure 44).
The tritium labelled synthesis of enalapril maleate (MK 421) has been accomplished by Bartroli et al. [147]. The methodology involved used N′-hydroxysuccinimidyl ester of N-carboxyalkyl alanine derivative and reacted with L-[4,5-3H] proline. The crude reaction mixture was treated with maleic acid to give the maleate salt of tritium labelled enalapril in 27% yield.
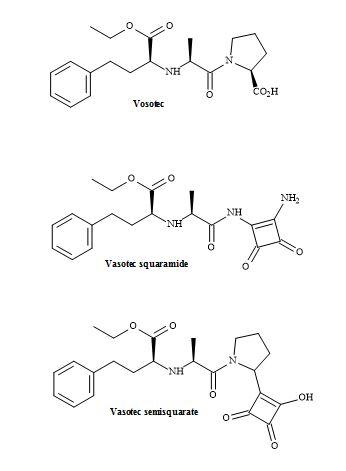
Figure 44. Vasotec and squaryl vasotec.
Peptidomimetics
There is a tremendous amount of current interest in the study of dipeptide analogues [148] shown in Figure 45. These isosteres of natural dipeptides are proving valuable for producing mechanism based enzyme inhibitors and proteolytically stable peptides, both of which hold great promise as therapeutic agents [149].
The challenge is the transformation of a bioactive peptide into a non-peptide entity to reproduce, as closely as possible, the relevant 3-D topography [150]. The aim was to mimic the pharmacophore – specific spatial arrangement of elements such as charges, lipophilic and hydrophilic groups – that are essential for recognition and interaction with the complementing domain of the receptor. This application leads to the introduction of the biochemical message, as in the case of agonists, and inhibition of antagonists [151].
The substitution of a peptide (CO-NH) bond for a squaryl entity, generates a new biological function. Therefore, a new lead discovery in the rational design of peptide-based drugs is paramount [152]. Ketovinyl and hydroxyethylidene dipeptide metaphors, as well as heterocyclic and unnatural amino acids, are shown to be replacements for the peptide bond [153]. The peptide bond isosteres (Figure 46) provide the platform, for studying the mechanism-based enzyme inhibitors and proteolytic reactions, on the stability of the unnatural peptide linkage. These isosteric analogues are important because they incorporate the key structural features of the known trans-double bond. Both ketomethylene and hydroxyethylene isosteres have the potential for directing alkylating agents (indicated by the arrows in Figure 46) such as cysteine thiol [153].
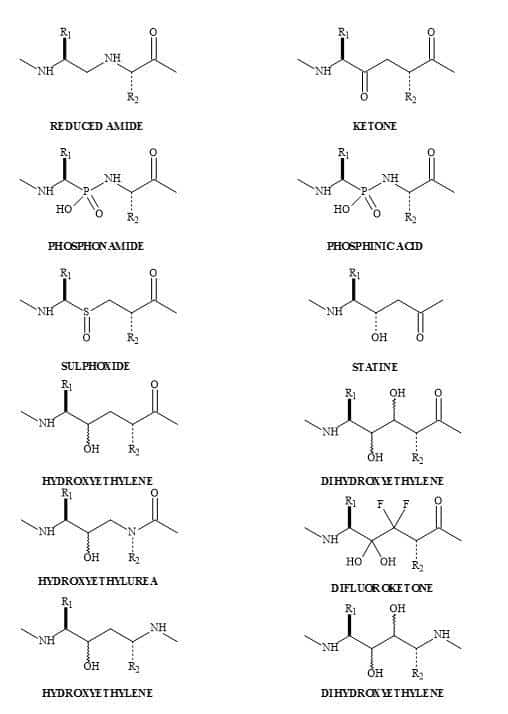
Figure 45. Dipeptide metaphors.
The hydroxyethylidene template can provide a tool for drug design, utilising the conformational restriction about the C-2 and C-3 bond. This method has been used to prepare angiotensin converting enzyme inhibitors based on the benzoyl-Phe-Gly-Pro [154]. Other similar templates function as statin mimics and have been made use of in structural activity studies on pepstatin-renin inhibitors [155].
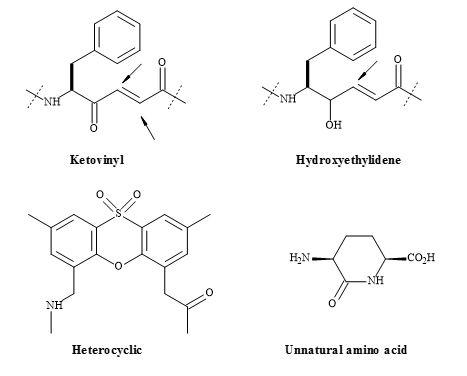
Figure 46. Ketomethylene and hydroxyethylene isosteres.
The biological activities of peptides are most often dependent upon primary and secondary structure [156]. The introduction of geometric changes into the peptide may well induce a different biological function. A squaryl molecular metaphor for the substitution of the peptide bond (RCONHR’) in established drugs, can lead to a whole array of new lead compounds, in the rational design of peptide hormones and neurotransmitters (Figure 47) [157].
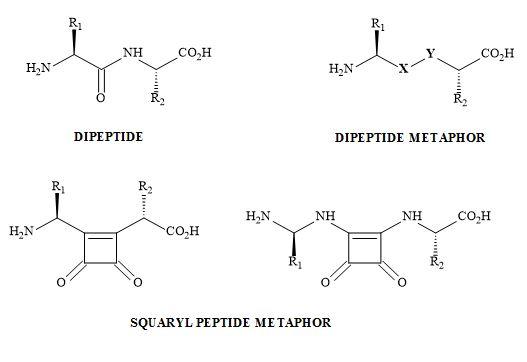
Figure 47. Peptide bond squarate metaphor.
Bisquaryls result from the coupling of halocyclobutenediones with squaryl stannane derivatives [158]. These reactions are catalysed with copper(I)iodide to assist in the palladium coupling of the two semisquarate derivatives. This technique is used when applied to the synthesis of squaryl peptides. The en route to bisquaryls derivatives utilises the technology developed by Liebeskind [132] shown in Figure 48.
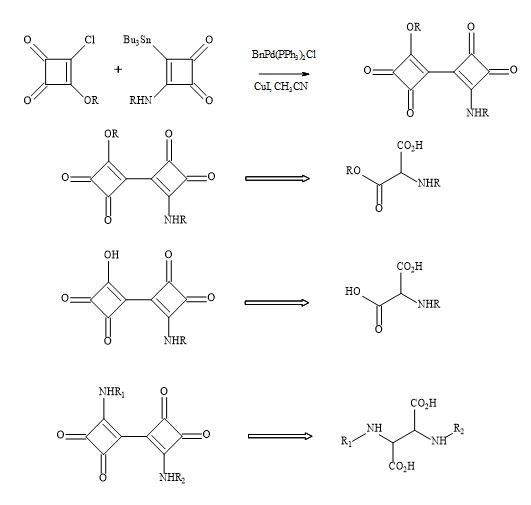
Figure 48. Bisquaryl peptide metaphors.
This technology generates a broad range of biologically active peptides and provides an understanding of the scope and limitations of the approach. Spectroscopic and theoretical conformational analysis give an insight into the effects of such modifications on conformational preferences [159]. The information gained will enable analogues of biologically active peptides to be synthesised and studied.
Aza dipeptides
Malaria causes over 800,000 deaths per year and is the most lethal of the Plasmodium species [160]. The infectious agent in humans is the Plasmodium falciparum and has acquired resistance to the majority clinically used antimalarial drugs [161]. In order to help in the search for antimalarial treatments, the public chemical library of GSK included four compounds containing a squaryl moiety, with activities ranging from 62 nM to 878 nM against the P. falciparum strain 3D7 [162].
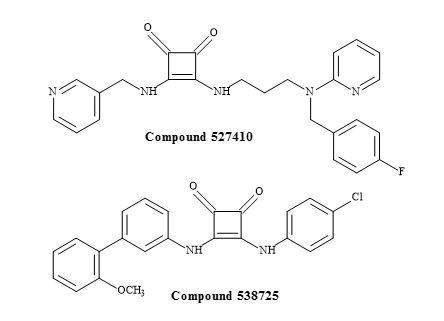
Figure 49. Squaramides from the GSK library.
The two squaramides in Figure 49 are antagonists of the serotonin receptor and were not an obvious choice in the rational drug design towards the malarial genome [163]. Extensive structural-activity relationship data was required to investigate these unusual squaryl moieties in the development of novel antimalarial pharmaceuticals.
However, this strategy led to the synthesis of aza squaryl derivatives, by using the methodology in Figure 50. An approach was to design a versatile scaffold by incorporating the recognition part at the double bond in the falcipain inhibitors [164]. A series of compounds were synthesised to contain the aza dipeptide recognition seaction, where the squaryl moiety acts as a replacement of the P1 carbonyl group. The amino acid residues chosen for these replacements included glycine, phenylalanine and homophenylalanine. The incorporation of the recognition seaction is a result of N-BocLeu and the corresponding hydrazine. Several squaryl derivatives showed increased antiplasmodial activity against CQ-resistant P. falciparum strain with negligible cytotoxicity. Overall, aza dipeptide squaryl derivatives, are promising lead compounds for the development of novel agents against P. falciparum malaria.
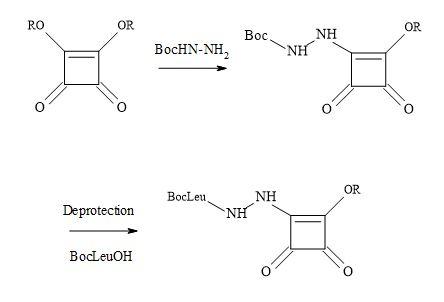
Figure 50. Synthesis of aza squaryl derivatives.
Squaramide lead compounds
The squaryl molecular metaphors continue to play a vital role in rational drug design to develop new treatments for various disease states. Squaramides have found use in the design of novel organocatalysts such as Rawal’s catalyst [165]. This squaramide (-)-cinchonine catalyst is utilized in the conjugate addition of 1,3-dicarbonyl compounds to β-nitrostyrenes, to obtain products with high entioselectivities. This bifunctional catalyst probably operates in a similar mode to Takemoto’s catalyst which contains a thiourea functionality [166,167].
The functionality of squaramides has the ability produce biologically active compounds. Squaramide is the conjugate base of squaric acid and can recognize the ammonium cation through its remarkable hydrogen bond accepting character, to produce a di-O-protonated squaramide. Consequently, this is due to the aromaticity of the four-membered ring in the complex form. A theoretical study has shown that squaramide complexed with ammonium cation is more aromatic in behaviour than squaramide and this maybe the result of the hydrogen bond acceptor character of squaramide (Figure 51) [168].
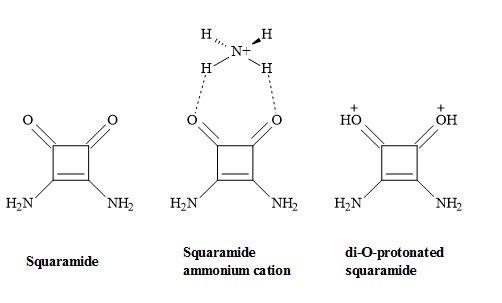
Figure 51. Squaramide complex.
Squaramides can mimic ureas, guanidines and phosphates including other reported functional groups. They can act as hydrogen bond acceptors and hydrogen bond donors towards cation and anion recognition sites of amino acids [169]. These compounds possess the vital parameters towards the design of antibody-drug conjugates (ADCs) [170] and radionuclide-antibody conjugates (RACs) [171].
Squaramides can selectively bind protein kinases including the CXCR2 receptor. Pharma are developing various CXCR2 selective antagonist receptors containing a squaramide metaphor, which has the potential to become a first-in-class anti-inflammatory treatment for chronic obstructive pulmonary disorder [172,173]. In addition to the squaramide CXCR2-CXCR1 dual inhibitors (Figure 52) [174].
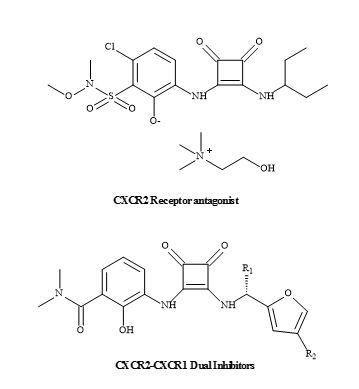
Figure 52. CXCR2-CXCR1 inhibitors.
Moreover, squaramides have been evaluated for antitumour activity in gastric carcinoma cells (HGC-27) shown in Figure 53 [175].
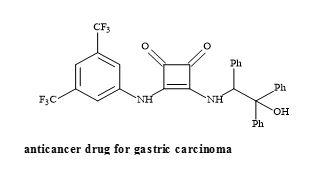
Figure 53. Structure of squaramide HGC-27.
Another interesting drug lead is the NMDA antagonists known as EAA-090 [176]. This NMDA antagonist possesses a squaramide in place of the α-amino acid functionality [131]. The squaryl moiety is structurally similar to α-amino acids – but is deficient in the general properties of amino acids – such as the basicity, acidity and nucleophilicity (Figure 54).
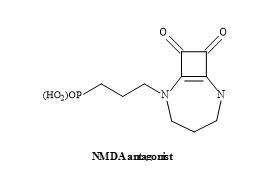
Figure 54. Structure of squaramide EAA-090.
Studies have demonstrated that EAA-090 is potent against the standard NMDA antagonist, CGS-19755 (Selfotel) [177]. These binding studies conducted in the presence of [3H]CPP using various rat brain models [178].
The research of Chi-Wan Lee et al. has discovered a novel peptidomimetic, containing the squaramide metaphor as the replacement of the guanidine group [179]. The squaryldiamide derivative was tested as a transactivator of transcription (Tat)-TAR inhibitor and was able to bind TAR RNA with high affinity. Therefore, the squaramide could be used as a potential metaphor of unsubstituted guanidine in peptidomimetics. Furthermore, applying RNA-targeted structure-activity-relationship (SAR) studies a novel peptidomimetic squaryldiamide was revealed to be a metaphor for guanidine. This metaphor was able to bind TAR RNA with a high affinity for the HIV type-1 tat protein (Figure 55) [180].
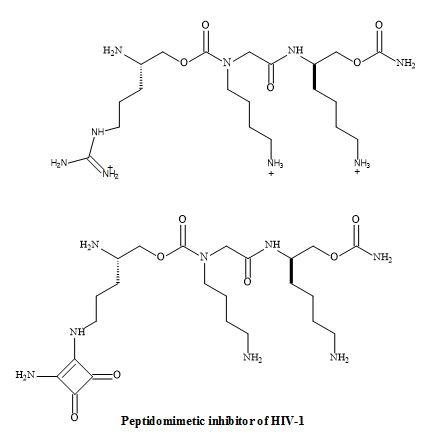
Figure 55. Squaryl peptidomimetic inhibitor of HIV-1.
Squaramides have been used to design a novel series of adenosine 5’-triphosphate-sensitive potassium (KATP) channel openers for the treatment of urge urinary incontinence (UUI) [181]. Furthermore, these studies identified that the N-cyanoguanidine moiety of pinacidil can be replaced with a squaramide to give the analogue (Figure 56) [112].
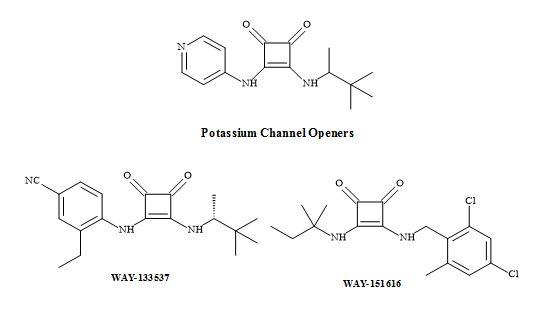
Figure 56. Potassium channel openers.
The squaramides WAY-133537 [182] and WAY-151616 [113] are claimed to have bladder selective properties over cardiovascular parameters in animal models (Figure 56). WAY-151616, indicated excellent tissue selectivity for bladder K channels over arterial tissue (MAP ED20 = 100 mg/kg; selectivity: MAP ED20/bladder ED50 = 166) [183]. These squaryl drug leads provide a novel class of KATP channel openers with unique selectivity for bladder smooth muscle in vivo. Further modification is required to produce the desired oral selectivity. However, SAR studies have shown that the incorporation of a squaramide as an amino acid metaphor, in a series of VLA-4 integrin antagonists, gave an improvement in the rate of clearance (Figure 57) [184].
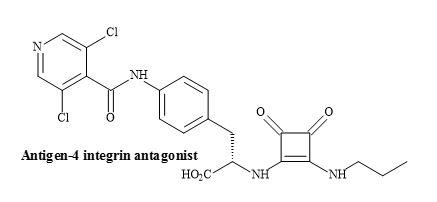
Figure 57. Squaramide VLA-4 integrin antagonist.
The effect of a novel histamine H2-receptor antagonist, pibutidine hydrochloride (IT-066) on the [3H]tiotidine binding to canine cloned H2-receptor-expressed hepa cells was evaluated. The squaramide, IT-066 displaced the binding of [3H]tiotidine to cells transfected with the wild-type H2-receptor in a concentration-dependent manner (Figure 58) [185].
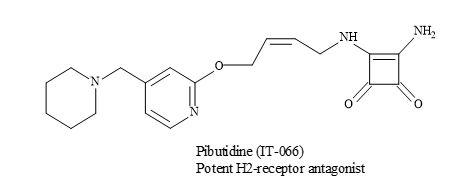
Figure 58. Pibutidine hydrochloride (IT-066).
These results show that IT-066 interacts and binds to the H2-receptor in a time-dependent manner. Consequently, the irreversible inhibition of IT-066 is important in the interaction with 190Thr especially in the fifth transmembrane domain of the H2-receptor [185].
Hybrid Antibiotics
The emerging and continued resistance to currently available antibiotics and the limited pipeline of new antibacterials urgently call for the development of new methodologies that can address the problem of growing antibacterial resistance [186]. The next generation of antibiotics may consist of hybrids containing structures based on fluoroquinolones, anthracyclines, oxazolidines, macrolides and others [187]. The advantages of such systems can produce a covalent binding that can make the pharmacokinetic characteristics of these molecules more reliable and improve the penetration of each component into the cell [188,189].
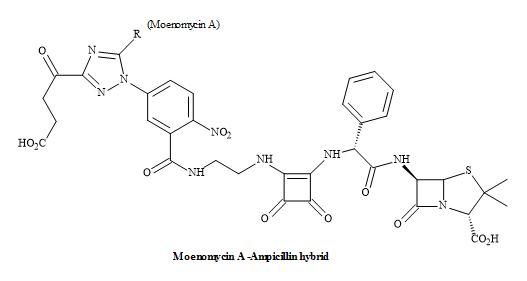
Figure 59. Squaramide hybrid antibiotic.
Consequently, the drug efficacy will increase due to inhibition of two targets but also the resistance to one or both antibiotics can be circumvented. The structures can be grouped together according to the type of covalent spacer (cleavable or not) connecting the moieties of two agents. Dual action antibiotics with a spacer/chemical linker that can be cleaved in living cells are known as dual action prodrugs [190].
However, the dual-action hybrid antibiotics mainly consist of two antibiotics that inhibit different targets in a bacterial cell. The possible advantages include activity against drug-resistant bacteria, increasing the spectrum of the activity and reducing the bacterial resistance (Figure 59) [191].
Access to a range of pharmaceutical drugs is a major concern for developing countries [192]. Consequently, Chagas disease which is caused by Trypanosoma cruzi is a typical example of a chronic disease without an effective treatment [193]. Current treatments were based on benznidazole and nifurtimox [194]. Unfortunately, these treatments proved to be expensive and have toxic side effects [195].
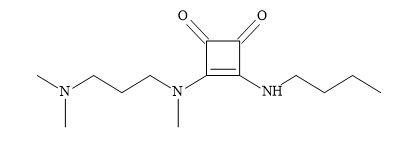
Figure 60. Anti-Chagasic agent.
The application of squaramides owing to the fact they are capable of functioning as hydrogen bond donor and acceptor groups. These properties enable multiple interactions with receptor sites. The incorporation of amine and carboxylic acid functional groups enable the squaramide compounds to have increased solubility and therefore, make them a suitable choice for the rational design of drugs. The authors applied the Lipinski’s rule of five [20] in the design of the squaramide candidates for treating Chagas disease [196]. Various studies confirmed that the squaramide compound (Figure 60) to be the most effective for both acute and chronic phases. The activity, stability, reduced cost of starting materials and straightforward synthesis make amino squaramides appropriate molecules for the development of anti-Chagasic agent [197].
Vitronectin receptor antagonists have a possible role in the treatment of restenosis following balloon angioplasty [198]. Urbahns et al. have investigated several novel arginine mimetic scaffolds leading to the insight of the alpha(V)beta(3)/ligand interaction [199]. Squaramides have shown that subnanomolar alpha(V)beta(3) antagonist with improved potency on human smooth muscle cell migration (Figure 61).
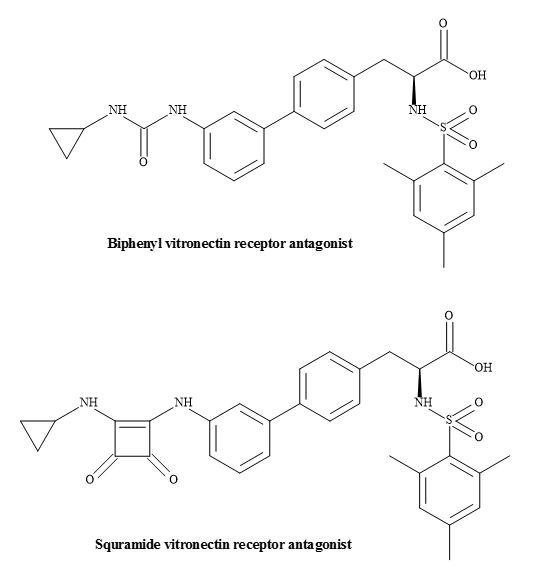
Figure 61. Vitronectin receptor antagonists.
The design and synthesis of a novel 3-hydroxy-cyclobut-3-ene-1,2-dione derivative were evaluated in the thyroid luciferase receptor assay. The substrate 3-[3,5-dichloro-4-(4-hydroxy-3-isopropylphenoxy)-phenylamino]-4-hydroxy-cyclobut-3-ene-1,2-dione has demonstrated selectivity towards thyroid hormone receptor β (Figure 62) [200].
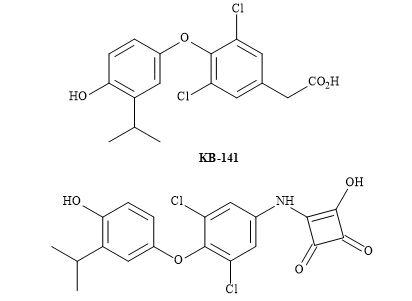
Figure 62. Squaryl of KB-141.
A rational structure-activity relationship for 3-hydroxy-cyclobut-3-ene-1,2-dione compound as a thyroid hormone receptor β indicated the squaramide to exhibit similar selectivities for TR-α and TR-β as that shown by KB-141. The more elaborate SAR, lead optimization, in vitro, in vivo studies and pharmacokinetics in rodent models, is required.
HIV-protease inhibitors
Polypeptides are regulators of life in biological systems [201]. These regulators are specific enzymes consisting of long chains of proteinogenic amino acids and facilitated by the transcription of RNA. At this stage, they are not in their final biological information form, and the peptide needs to modify to the correct length. Subsequently, this is accomplished by using nature’s molecular scissors, known as proteases [202]. They merely cut the chain at specific locations so as to transform the enzyme to its active form.
The shape of HIV is spherical with a diameter of 0.1 micrometres and the outer viral envelope consists of two layers of lipids. These lipid membranes are derived from a human cell, when a newly formed virus particle buds from the cell [203]. The embedded viral envelope contains proteins from the host cell, containing approximately 72 copies of a complex HIV protein that projects from the envelope surface. This protein is known as Env and consists of a cap made of three or four molecules called glycoprotein (gp)-120. Also, the stem consists of three or four gp41 molecules that anchor the structure in the viral envelope. The envelope proteins are a focus in the development of HIV vaccines [203].
Renin is an essential protease enzyme, which has received much attention in its mode of action. The central role of this polypeptide is to cut the larger peptide angiotensinogen, to give the corresponding decapeptide angiotensin I. Angiotensin I, initiates the potent precursor vasoconstrictor octapeptide angiotensin II on release with another protease called angiotensin converting enzyme (ACE) [204].
The mode-of-action of proteases is to stabilise a relatively high-energy transition state. In this case, the initial adduct is the hydroxy centre or its oxidised equivalent, the carbonyl carbon. This transformation causes the geometry at the centre to change from sp2 to sp3 [205]. Established inhibitors, consist of part molecules, that mimic a crucial stretch of the protease recognition site and most importantly provide a sequence that duplicates the transition centre sterically without including a cleavable bond [206].
The fermentation product pepstatin is a peptide-like inhibitor of pepsin and offers an insight into the rational drug design of the protease inhibitors. The central position of pepstatin provides a 1,3-hydroxyamide sequence. The 1,3-hydroxyamide is thought to act as a transition state analogue to the peptide bond. Note that a methylene replaces the single amide nitrogen. The active moiety, statin, has been prepared by the total synthesis involving aldol-like condensation of isoleucyl aldehyde with the lithio carbanion from acetate [207] shown in Figure 63.
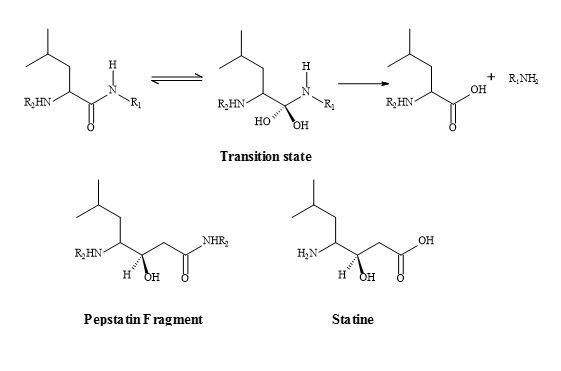
Figure 63. Peptide bond transition state analogue.
The α-hydroxy-β-amino acids are well-recognized main components for a variety of protease inhibitors and offer a new generation of pharmaceuticals. In many instances, the bioactivity of α-hydroxy-β-amino acids is expressed in the inhibition of aspartyl protease (renin, HIV protease). This mode of action is the ability of amino acids to mimic the tetrahedral intermediate in the hydrolysis of proteins. The process mediated by the aspartyl proteases, results in a blocking of the enzymes [207].
The functional simplicity of viruses, combined with the fact that they require a living host for their replication, has made them an unusually problematic therapeutic target [207]. Human immunodeficiency virus (HIV), in common with most viruses, consists of a packet of genetic information encoded in this instance in RNA and an outer protein coat. One of the final steps in viral replication involves the synthesis of the coat peptide. Production of this coat peptide involves scission of the initially produced much larger protein using aspartyl protease; a virus lacking the correct coat is not functional. This kind of thinking has led to the research of designer protease inhibitor drugs [208].
HIV-protease inhibitors are promising chemotherapy agents of HIV infections [209]. The HIV protease functions as a C2 symmetric homodimer [210] as shown in Figure 64. By applying the concept of mimetism to the natural substrate of HIV, to replace the interaction sites of the enzyme, novel peptide-based inhibitors of HIV-1 proteinase have been designed with C2 symmetry [211].
These are analogues that resemble the TS for the natural polyprotein substrate used by the HIV-protease, as the late stage in the replication of HIV. The β-amino acids are incorporated as a central building block in bis-symmetric lipophilic side chain arrangements. These derivatives showed high inhibition activity of HIV-protease [212].
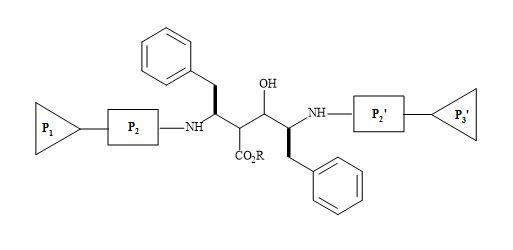
Figure 64. C2 symmetric homodimer.
Protease inhibitors are antiviral drugs. The mode-of-action is to interrupt the way that HIV utilises healthy cells to make more of the virus. When HIV enters a cell, its only goal is to make more viruses to infect other healthy surrounding cells. HIV hijacks the cell, to produce certain proteins the virus can use to copy itself. These proteins, utilised by the virus, include reverse transcriptase and protease. The objective of protease inhibitors is to stop the protease from helping to assemble a new viral form [213].
The FDA has approved the following protease inhibitors [214]: atazanavir (Reyataz, 2003), darunavir (Prezista, 2006), fosamprenavir (Lexiva, 2003) and indinavir (Crixivan, 1996). In addition to lopinavir/ritonavir (Kaletra, 1996), nelfinavir (Viracept, 1997), ritonavir (Norvir, 1996) and saquinavir (Invirase, 1995).
HIV-protease is an important target for drug design [215]. There are several high-resolution X-ray crystal structures available both of the protein alone and also complexed with a variety of inhibitors [216]. Numerous HIV-protease inhibitors have been in development over the last few decades for their biological and pharmacological properties [217]. For example, amprenavir derived from the prodrug fosamprenavir is an aspartyl protease inhibitor, containing a (2R,3S)-1,3-diamino-2-hydroxy-4-phenyl-butane (in bold) as the key subunit [218] as shown in Figure 65.
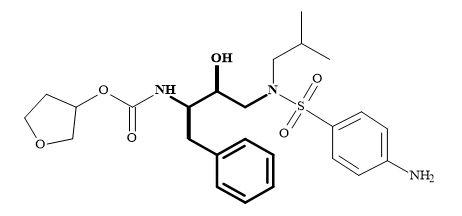
Figure 65. (2R,3S)-1,3-Diamino-2-hydroxy-4-phenyl-butane subunit embedded in amprenavir.
A possible lead modification to the aspartyl protease inhibitor family is to incorporate squaryl entities. These incorporated squaryl moieties can then interact probably by hydrogen bonding, at the active site, in the HIV core: namely with the two aspartic acid residues, one fully ionised and the other not [211]. This interaction will then ‘gum up’ the aspartic acid molecular scissors thus preventing the HIV making proteins for replication of itself (Figure 66).
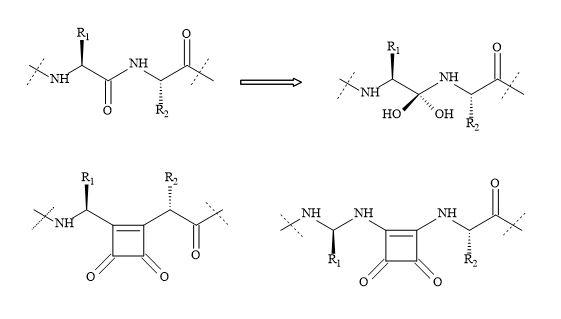
Figure 66. Squaryl group in protease inhibitors.
An interesting approach to the design of HIV protease inhibitors is from the research of the Dupont Merck group [219]. This new class of inhibitors are based on a cyclic urea core (Figure 67).
Figure 67. Squaryl HIV inhibitor.
The uniqueness of this class of inhibitors is the cyclic urea carbonyl oxygen that mimics the hydrogen bonding features of the key structural water molecule [211]. The premise behind the incorporation of the cyclic urea core into an inhibitor would be to replace the flap water molecule, which could lead to improvement in binding energy. This positive entropic effect should be provided by the cyclic urea core.
This class of inhibitors also contained the diol functionality as a transition state mimic to interact with the catalytic aspartates. Several inhibitors based on these concepts were developed and found to be potent against HIV proteases [220]. To date, these inhibitors have not yet been approved for HIV therapy.
The squaryl HIV drugs may be simpler to manufacture than the already known inhibitors. Therefore, lowering the drug price and allowing poorer nations to access these new squaryl designer drugs would be of great benefit.
Glutathione
The non-proteogenic amino acid S-buthionine-S,R-sulphoximine (BSO) is a potent glutathione inhibitor under investigation as a potential chemotherapeutic agent for the treatment of various cancer states such as malignant melanoma (Figure 68) [221]. Specifically, the S,S-BSO is used to deplete glutathione, the main protectant compound in tumours. Therefore, this permits the subsequent use of an anticancer (antineoplastic) agent or radiation to be more effective at a lower dose.
Moreover, it has been proven that S,S-BSO is more active as a chemotherapeutic agent at even lower doses. Biological studies have shown that S-buthionine-S-sulphoximine (S,S-BSO) is more potent in the inhibition of the tripeptide glutathione than S,R-BSO [222].
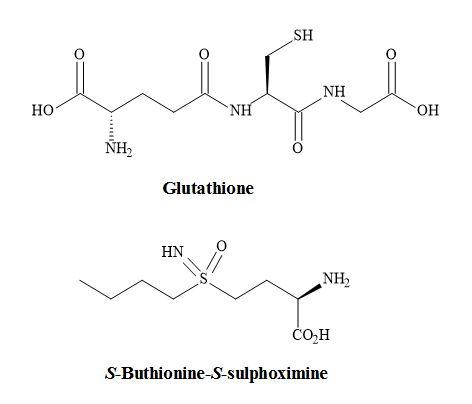
Figure 68. S-Buthionine-S-sulphoximine.
Figure 69 demonstrates a potential strategy to develop the next generation of glutathione inhibitors. The carboxylic amino moiety in S,S-BSO could be substituted for the squaryl entity 3,4-diamino-3-cyclobutene-1,2-dione. The squaryl metaphor contains a dipole and therefore makes it an electronic mimic of the ammonium carboxylate; equivalent to the amino acid moiety in S,S-BSO.
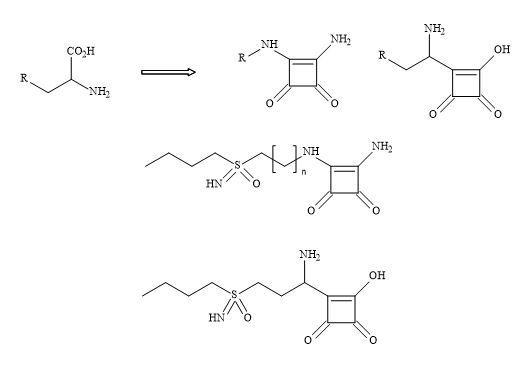
Figure 69. Squaryl buthionine sulphoximine.
This subtle substitution converts the chiral centre of the amino acid to give an achiral compound but leaving the chirality at the sulphoximine unchanged-namely the Schiral centre.
Histone deacetylase inhibitors (HDAC)
Several groups have prepared analogues of suberoylanilide hydroxamic acid (SAHA) by replacing the zinc binding moiety with N-hydroxyurea and 4-hydroxymethyl oxazoline units [223]. The application of squaryl entities provides the platform to chelate the biological zinc (Figure 70). The aliphatic carbon chain can be varied to optimise the pharmacokinetics of these novel HDAC inhibitors.
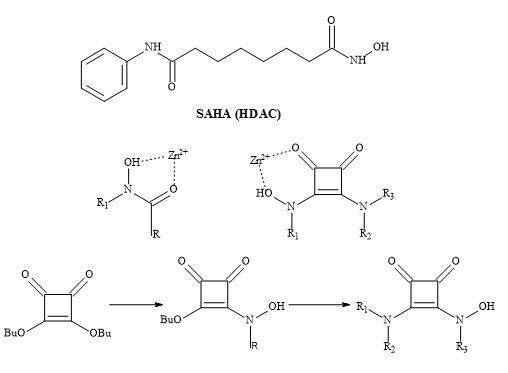
Figure 70. The binding mode of hydroxamic acid and squaric acid N-hydroxylamide amide.
The therapeutic class histone deacetylase inhibitors are central to the design neurological and psychiatric drugs [224]. A series of suberoylanilide hydroxamic acid (SAHA) substituted compounds was synthesised and evaluated for their inhibitory and cytotoxic activity.
Metalloprotease inhibitors
Matrix Metalloproteases (MMP’s) help cells spread and set up new blood supplies to support tumour growth. By designing a selective inhibitor that gums up the enzyme and reducing the spread of cancer [225].
The interaction of typical inhibitors is between the sulphone and the episulphide with the residual amino acids (leu-191 and ala-192) is facilitated by the zinc ion centre in the metalloprotease [226]. The inhibitor then becomes covalently bound to an active site glutamate residue (glu-404). The substitution of the sulphone moiety in the inhibitor below for a squaryl entity may produce a better metalloprotease drug. The squarate derivative shown in Figure 71 may react with the residual amino acids in the protein and therefore stop the process of metastasis [30].
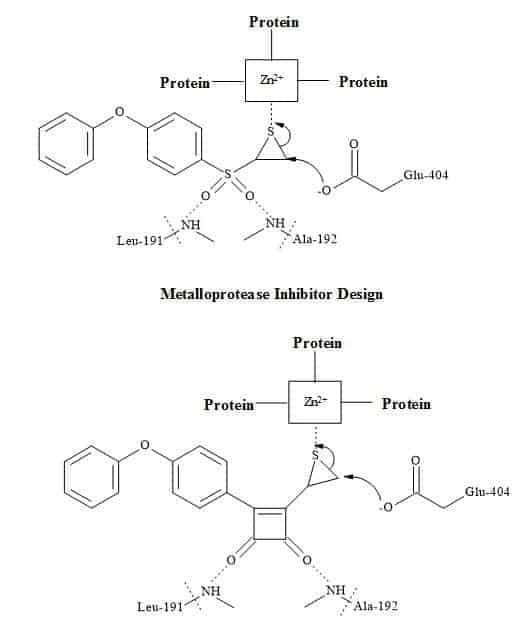
Figure 71. Matrix metalloprotease metaphors.
Anti-migraine drugs
Anti-migraine drugs can generate vast revenues for the pharmaceutical sector. Figure 72 shows the chemical structures of the leading commercial migraine drugs [227]. All of these drugs contain an indole pharmacophore for their therapeutic action on selective CNS agents [228,229].
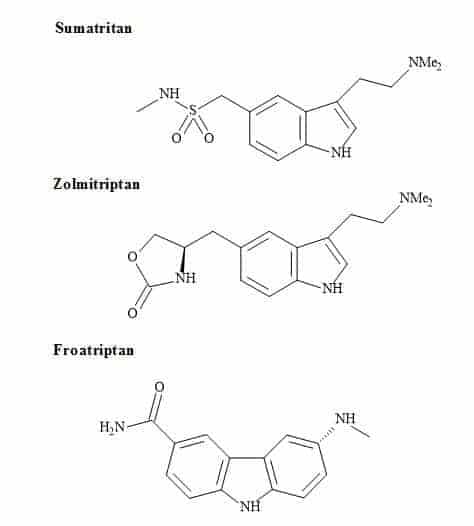
Figure 72. Antimigraine drugs.
The indole derivative, serotonin (5-hydroxytryptamine) is an important neurotransmitter in the brain [230]. Several mental health indications and in particular depression are linked to inappropriate levels of receptor sensitivity to this compound [231]. The serotonin moiety has reported in the design of CNS drugs in an attempt to ensure interaction with brain receptors [232]. A quite recently introduced compound that is effective against migraine, sumatriptan, bears a closer resemblance to serotonin [233].
BMS-181885 is a potent 5-HT1D agonist developed by Bristol-Myers Squibb [234]. It is one of several second-generation triptans under development for the treatment of migraine. BMS-181885 is structurally and pharmacologically similar to that of GSK drug, sumatriptan.
The sulphonamide moiety in sumatriptan is a non-classical metaphor for the carboxylic acid. Therefore, molecular metaphor principles can be replaced with a squaryl entity (Figure 73).
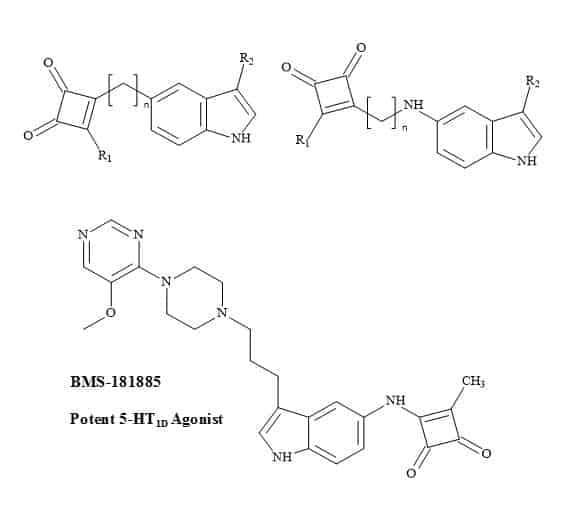
Figure 73. Antimigraine squaryl metaphors.
IMDH enzymes
The biochemistry of Thermus thermophiles β-isopropylmalate dehydrogenase (IMDH), the key enzyme of the leucine biosynthesis in yeast, bacteria and higher plants occurs in two simple transformations (Figure 74) [235]. In the first step (2R,3S)-isopropylmalate is oxidatively decarboxylated to give ketoisocaproate and secondly the leucine keto acid is then subsequently transaminated [236].
The intermediate in these two steps is the decarboxylation to give an enol or enolate that undergoes stereoselective protonation. Squaryl derivatives can act as enol/enolate substrates in the inhibition of the IMDH enzyme. Due to the constraint of the cyclic structure in semisquarates they can only function as (Z)-enolates [55].
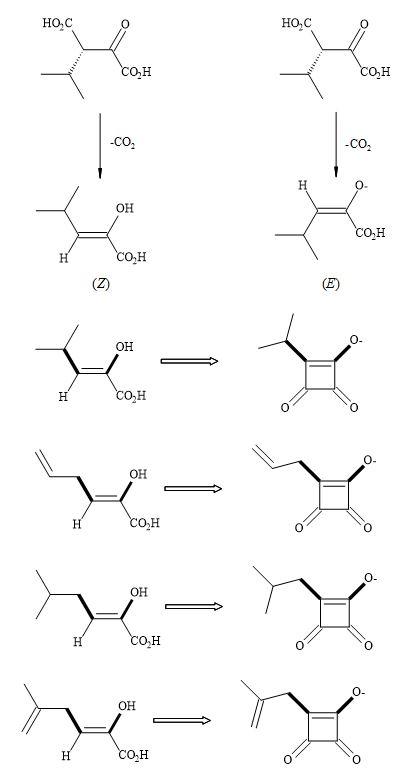
Figure 74. Thermus thermophiles β-isopropylmalate dehydrogenase (IMDH) – squaryl analogues.
Phosphonate and squarate analogues
The American Cancer Society has projected for 2017 the total number of cancer diagnoses to be 1,688,780 and 600,920 cancer deaths in the US. Cancer accounts for 25% of all deaths in the USA. According to the National Institute of Health, over $40 billion was spent annually in direct medical costs to treat cancer patients in the USA [237]. Therefore, there is a need for new cancer treatments that are both therapeutic as well as cost-effective [238].
Several research groups have synthesised a series of novel anti-cancer agents with a glycerol backbone or aliphatic chain structure linked to phosphorus-containing side chains, such as phosphonocholines (phospholipids) and phosphonoglycerols [239]. Platelet Activating Factor (PAF) is a phospholipid composed primarily of the C16 and C18 homologues [240]. The biological properties are its potent ability to aggregate platelets and to lower blood pressure [241]. These novel phosphonates may be lead compounds to develop squaryl drugs targeted towards the treatment of lung, breast, colorectal, cervical and ovarian cancer, as well as lymphomas, leukaemia, and melanomas. Squaryl molecular metaphors continue to play a role in the rational drug design of anti-inflammatory, anti-allergy and anti-cardiovascular disease properties and potential treatment for arthritis, asthma and hypotension [96].
The general approach to squaryl metaphors is the substitution of the phosphonate for a molecular entity in which the carbon to oxygen bond in the phosphate monoester is substituted by a stable carbon to carbon bond of the squarate (Figure 75). This squaryl for phosphate substitution can open a broad range of drug candidates, which may survive the deactivation and elimination process before the drug substance can be delivered and therefore produce effective treatments.
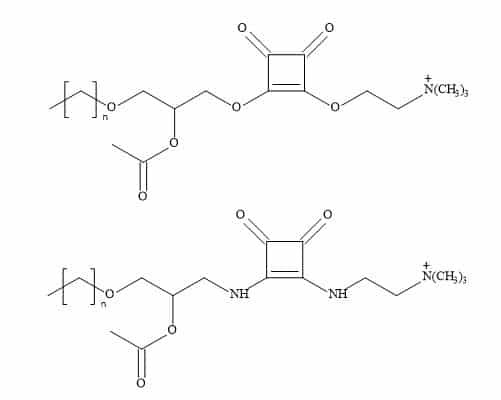
Figure 75. Squaryl for phosphate substitution.
Wissner et al. have prepared a host of structural analogues of this vital substance [242]. These include the replacement of the phosphocholine portion of the molecule with a neutral sulphonyl bismethylene group (Figure 76). Moreover, a squaryl-phosphonate syllogism can be applied directly to the phospholipid PAF structure to give the potential squaryl analogue.
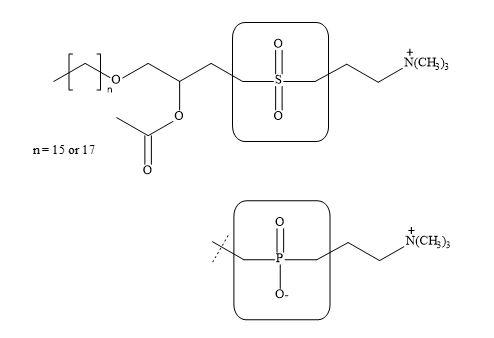
Figure 76. Sulphonyl bismethylene group.
The authors have demonstrated that the sulphonyl bismethylene moiety in the PAF structure resulted in a decrease in biological activity. Literature precedents have indicated that the sulphonyl bismethylene group has similar steric requirements to the phosphate group. Since these two groups differ in electronic characteristics, it may well be the ability of the phosphate group to bear a negative charge that is responsible for its contribution to the activity of PAF [243].
Medicinal drugs undergo various detoxification processes in the human body. These processes may involve the potent nucleophile glutathione. This tripeptide interacts with the drug and makes it less pharmacologically active [244]. Other mechanisms are in place which can interfere with the drug candidate namely glucuronidation [245]. The glucuronidation process involves the conjugation primarily with hydroxyl, carboxyl, amino, heterocyclic nitrogen and thiol groups [246]. If these functional groups are absent in the drug substrate, they can be introduced directly by chemical methods.
The glucuronidation process is not simple and involves the use of the activated derivative of glucuronic acid, uridine diphosphate glucuronic acid (UDPGA). UDPGA is synthesised in a two-stage process starting from glucose-1-phosphate and is first coupled to uridine triphosphate to give UDP-glucose. UDP-glucose undergoes oxidation by the enzyme UDPG-dehydrogenase which uses NAD as a cofactor to produce UDPGA [247].
Nucleoside diphosphate sugars are intermediates that donate glycosyl residues in the biosynthesis of polysaccharides, glycolipids, glycoproteins and some components of the bacterial cell wall. Compounds that interfere with protein glycosylation show a variety of biological effects, such as inhibition of the replication of enveloped animal viruses [248].
For instance, 2-deoxy-D-glucose is a glycosylation inhibitor having antiviral activity, converts to UDP-2dGlc and GDP-2dGlc and consequently interferes with the biosynthesis of the linked oligosaccharide precursor of the N-glycosylated glycoproteins [249]. Replacement of the phosphonates in the UDPGA with squarate entities may produce potent inhibitors for this enzyme and therefore enable protection against the administered drug from being eliminated before it can reach the specific target.
Glycobiology
Complex oligosaccharides can indicate a significant number of diverse and important biological functions. It has been a major incentive for devising new methods for the chemical and enzymatic synthesis of this class of molecules [250]. The introduction of squaryl molecular metaphors into this field of chemistry generates a new set of glycol squarates. Consequently, expressing different biological properties in the oligosaccharide [251]. Hence, the substitution of the phosphorus linkage with a squaryl entity provides a range of interesting oligosaccharides [252]. Moreover, squaryl coupling of carbohydrates bearing amino-terminus spacer arms to proteins (Figure 77).
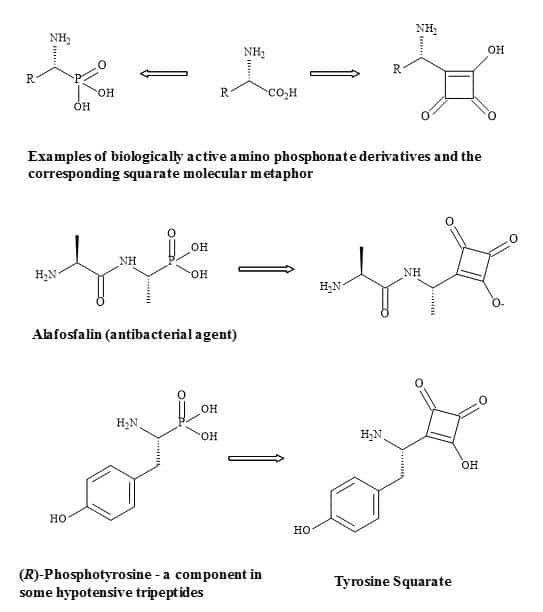
Figure 77. Substitution of phosphorus linkage with a squaryl entity.
Squaramides contain a four-membered ring system related to squaric acid and are capable of forming four hydrogen bonds. A high affinity for hydrogen bonding is a product of the increase aromaticity of the ring system. This hydrogen bonding and aromatic swapping, in combination with structural rigidity, has been exploited in many of the applications of squaramides [253].
The formation of squaramide derivative takes place under relatively mild or aqueous conditions. This approach makes them ideal units for bioconjugation and supramolecular chemistry [254].
The research group of Tietze demonstrated the conjugation of two amines with squarate ester [255]. The key feature of this reaction is the slight excess of amines to give high yields of the two amine components. UV spectroscopy can be used to analyse this process. For example, in the synthesis of glycoconjugates, where the cyclobutene-1,2-dione skeleton is linked to a saccharide, protein or immunogens [256]. In a recent work hydrophobic didecyl squarate was reported to be a reagent to handle with ease for purification, as shown in the conjugation of bovine serum albumin (BSA) (Figure 78). Recent developments in glycoconjugate technology can be applied for drug immunotherapy and also on a variety of oligosaccharide-squaramide-protein (e.g. bovine, human and chicken serum albumins) conjugates have been prepared with the Tietze’s squarate ester methodology and tested for their bioactivity [257]. Furthermore, a one-pot procedure was developed by Alegre-Requena et al. to give unsymmetrical squaramides that can generate large compound libraries [258].
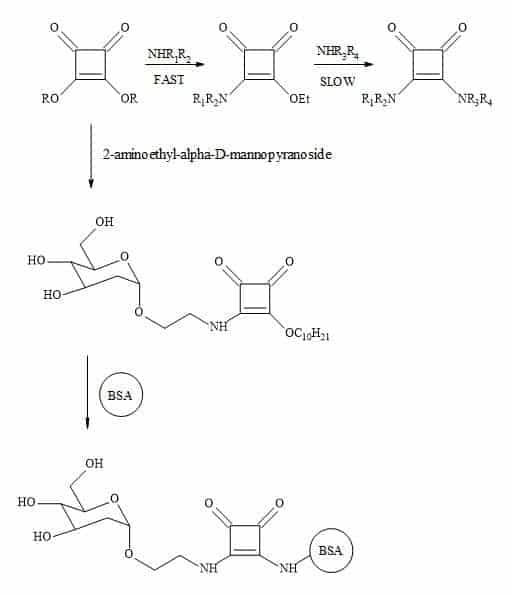
Figure 78. Tietze’s methodology towards squaramide conjugation with biomolecules.
α-Amino phosphonic acids
The α-amino phosphonic acids and cyclic α-aminophosphonic acids [259] and their esters serve as attractive substitutes for amino carboxylic acids in biological systems. The absolute configuration of the α-carbon strongly influences the biological properties of α-amino phosphonic acids [260]. The pKa values of α-amino phosphonic acids are similar to those of the corresponding αamino acids [261].
Phosphonic acids are doubly-ionized at physiological pH and may affect their potency as enzymes inhibitors. These compounds exhibit interesting and useful properties such as peptide analogues [262], antiviral agents [263] and haptens for the generation of catalytic antibodies [264]. This moiety is present in potent antibiotics [265], herbicides and pesticides [266].
When studying drug design, it is important to understand the Transition-State (TS) of the potential inhibitors. A transition-state mimic has the ability to bind an enzyme at its point of interaction as strongly as any available inhibitor. These strong binding interactions will prevent enzyme activity. In order to identify a particular inhibitor of the TS of an enzyme computational modelling and experimental techniques are used to reveal the geometric shape [267].
The introduction of squarate entities in TS theory may prove futile in the development of lead drug modification. Figure 79 shows an example of demonstrating how a squaryl TS analogue can replace phosphonate esters.
Glycopeptides
In 1999, the structure of FimH was determined by x-ray crystallography studies and found to comprise of two folded domains [268]. These studies indicated that FimH is a mannose-specific bacterial lectin found on type 1 fimbriae with a monovalent carbohydrate recognition domain (CRD). The N-terminal adhesive domain plays a key role in surface recognition, while the C-terminal domain is responsible for organelle integration. The two domains linked via a tetra-peptide loop, in addition to a carbohydrate-binding pocket are located at the N-terminal domain. Further studies have indicated that type 1 fimbrial adhesins are conserved in the core structure. Several studies have demonstrated that in vitro induced mutations can lead to the modification of the C-terminal domain [269].
Consequently this results in bacterial adhesion, forming dual bending sites and related binding phenotypes [270]. Furthermore, binding studies using multivalent ligands have indicated regions of the carbohydrate-binding sites on the protein. A squaramide moiety was used as a bivalent glycopeptide ligand and has the ability to bridge two carbohydrates on FimH. The bivalent glycopeptide, containing a squaramide shown in Figure 80, was synthesised to test this hypothesis of two carbohydrate binding sites on FimH protein [271]. The methodology involved reacting azido-functionalized mannotrioside and 2-azidoethyl mannoside with diethyl squarate. The authors concluded that further studies are required to test this hypothesis [271]
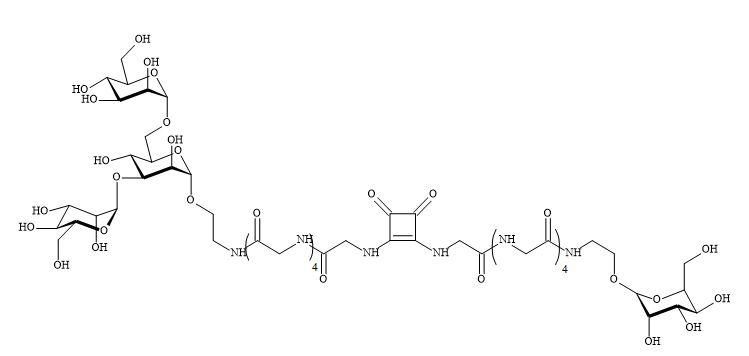
Figure 80. Bivalent glycopeptide squaramide.
Nucleic acid squarate analogues
The chemical modification of oligonucleotides has gained considerable interest due to their potential application as regulators of gene expression [272]. The Figure 81 below outlines the possible metaphors of the modification to the phosphodiester linkage.
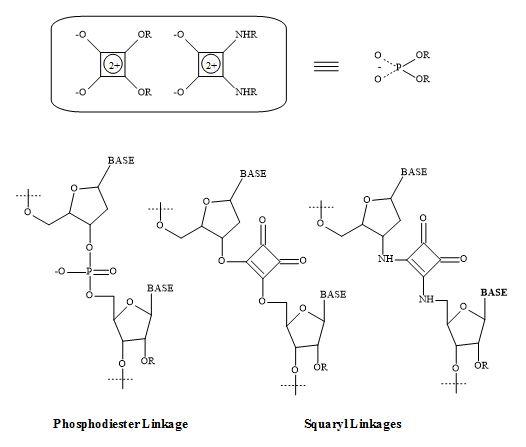
Figure 81. Phosphodiester linkage and squaryl metaphors.
The use of phosphonate analogues, such as biological phosphate monoesters has been governed primarily by the general stability of the phosphorous-carbon bond [273]. The oligonucleotides analogues, containing a squaramide linkage, were developed to mimic the phosphate group [273]. The modified thymidine dimer derivative T3’sq5’T which has a squaramide linkage was synthesised by a two-step substitution with diethyl squarate and incorporated into oligodeoxynucleotides.
The DNA duplex [5’-d(CGCATsqTAGCC)-3’/5’-d(GGCTAATGCG)-3’] was found to be distorted at the modified dimer site but preserved the base pairing ability. Continued work with U2’sq5’T indicated unique base-pairing ability for oligonucleotides with G at the site opposite to the T of U2’sq5’T. This research suggests the possibility to create a new class of antisense/antigene drugs and nucleotide analogues based on squaryl-nucleoside (base) conjugates [274].
The antineoplastic agent, 6-mercaptopurine produces a 50% remission rate for childhood acute leukaemia [275]. This substrate inhibits several enzymatic systems, but these inhibitions are irrelevant to its anticancer activity. Only tumours that convert the drug to its nucleotide are affected [276].
The immunosuppressive drug 6-mercaptopurine underwent bioactivation by a reaction with 5-phosphoribosylpyrophosphate and catalysed by hypoxanthine-guanine phosphoribosyl transferase (Figure 82). The final substrate inhibits several enzymes in the purine nucleotide biosynthetic pathway. However, the most prominent site is one of the first enzymes in the de novo pathway, namely, phosphoribosylpyrophosphate amidotransferase and catalyses the conversion of phosphoribosylpyrophosphate to phosphoribosylamine [277].
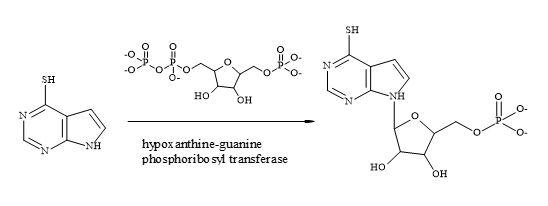
Figure 82. Bioactivation of 6-mercaptopurine.
The introduction of squaryl entities into nucleoside-based drugs such as anti-cancer and antiviral may prevent these drug substances from being deactivated by the enzymes that take part in the elimination processes. These squaryl nucleoside drugs may have a higher reactivity (more potent). Therefore, a low dosage relationship is required and may eventually lead to fewer side effects [278]. The squaryl analogue of cytidine 5′-monophospho-3-deoxy-D-manno-2-octulosonic acid 3-deoxy-D-manno-2-octulosonic acid (KDO) is a vital component of the outer membrane lipopolysaccharide of all Gram-negative bacteria (Figure 83). Several groups have pursued inhibition of KDO metabolism as a strategy for the development of novel anti-infective agents [279].
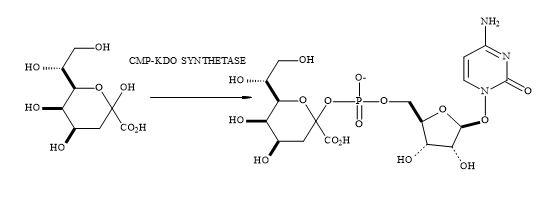
Figure 83. Synthesis of KDO.
PET imaging
Positron emission tomography (PET) imaging is a recognised diagnostic tool used in nuclear medicine to obtain clinical information from the patient. An extension of this imaging technique is the PET-CT hybrid. This system consists of two scanning machines, namely the PET scanner and an X-ray computed tomography (CT) scanner. These modern medical modalities occupy a central role in the technological apex of nuclear medicine and provide a prominent role in theranostics [280].
The second hybrid is a PET scanner combined with magnetic resonance imaging (MRI). This powerful imaging tool can obtain clinical images and provide corresponding information about the diagnosis of many disease states. These hybrid scanning machines are used in the clinical setting to show the effect of current and new therapies. PET scanning can detect the in vivo processes by which certain cancers utilise sugar molecules; while MRI provides for example structural details of tissues within a brain tumour. The major advantage of using PET-MRI scanning over PET-CT is the two-fold reduction in radiation dosages given to the patient. PET-MRI is a proven valuable imaging tool used in the areas of oncology, neuropsychiatric and heart diseases [281].
Consequently, PET-CT scanning can evaluate diseases via a functional approach, including morphostructural analysis. The benefit of using PET hybrid scanning is the potential for early diagnosis of the disease state. This method leads to a favourable prognosis and personalised therapy for the patient. Today, the most utilised PET radiotracer, currently used for oncology is [18F]fluorodeoxyglucose (FDG). In 2015, the number of clinical PET and PET/CT scans performed in the USA were expected to exceed 1.7 million. However, this is a net increase of about 6% compared to 2014. The scanning of patients takes place at 2,380 sites in the USA using fixed or mobile PET, PET/CT or PET/MR machines [282].
Vital diagnostic information can be gained using [18F]-FDG and several other radiopharmaceuticals. These include fluorine-18 florbetapir (Amyvid) [283], fluorine-18 florbetaben (Neuraceq) [284], fluorine-18 flutemetamol (Vizamyl) [285], fluorine-18 ethylcholine (FECH) [286] amongst others, with the overall aim to evaluate in vivo biological processes. PET imaging is creating numerous opportunities in the area of molecular imaging and provides a platform for a potential revolution in clinical theranostics especially in oncology, neurology and cardiology [287].
The history of PET imaging has been the focus of research in the human body since the 1970s [288]. This impressive non-invasive technological platform has seen a prominent clinical role in diagnosis, staging and monitoring of various disease states in patients since the 1990s. The pioneering research of Brownell and Sweet at the Massachusetts General Hospital in 1953 allowed for the first positron detector to study brain function in humans. Since then the imaging scientists Kuhl, Pogossian, Wolf, Sokoloff, Phelps, Di Chiro, Alavi and Wagner have made further contributions to the future development of clinical PET imaging [289].
In the 1990s, Wagner showed that the glucose analogue, FDG could be utilised in various PET studies [290]. Preliminary applications of clinical PET imaging were performed for the studies of brain disease, oncology and dementia. In the late 1980s, the advancement of technology brought about faster and more accurate whole-body examinations. These images were acquired by using PET-FDG scanning and become a valuable clinical tool in oncology. The central role of PET-FDG in nuclear medicine departments was for diagnosing, staging and re-staging of cancer in patients on a daily basis [291].
In 1997, the FDA gave approval for the regulation of PET radiopharmaceuticals used in diagnostic imaging [292]. Subsequently, PET-FDG became an established imaging technique in the clinical assessment of many neoplasms, finding a role also in non-malignant diseases for example dementia, myocardial ischaemia, inflammation and infection [280].
The fundamental aspect of PET imaging is the detection of radiation inside the human body. The radiation emitted is due to the decay of short-lived radionuclides. In the decay process, a positron (e+) and neutrino (v) are simultaneously emitted. The positron is an anti-matter electron (e–) with identical mass to an electron having a positive charge. In the decay process, the ejected positron decreases in kinetic energy by colliding with the surrounding atoms. This process causes the positron to come to rest. These positrons have characteristic energies peaking at 0.63 MeV and have a very short range within the tissue [293].
Figure 84. Positron-electron (e+ – e–) annihilation.
The gamma rays exit from the patient’s body and interacts with the scintillation crystals. These crystals are made of BGO (bismuth germinate), LSO (lutetium oxyorthosilicate) and GSO (cerium-doped gadolinium silicate). These, are also included LYSO (cerium-doped lutetium yttrium orthosilicate) extending to the photomultiplier tubes in the rotating detectors [294].
The positron hits an electron (e–) resulting in an annihilation reaction which results in energy. This energy is the product of two annihilation photons (511 keV), from the point of (e+ – e–) interaction in opposite directions. The annihilated photons are quantified using the principles of coincidence PET detection of gamma rays (Figure 84) [295].
These crystals convert gamma radiation into photon energy. The transducer converts the photons into electrical signals that are registered by the tomography electronics. The information generates 3-D real-time images, for example, the brain or a whole-body scan [296].
Imaging agents
The application of PET imaging is an excellent tool towards the development of novel medicinal drugs and improved clinical imaging. Therefore, a technological drive to generate novel radiotracers for a variety of in vivo biological targets is clearly becoming apparent [297].
To date, the fluorine-18 (18F) is the most employed PET radionuclide due to its manageable half-life of 109.7 mins, compared to carbon-11 (20 mins) which allows the commercial distribution of 18F-radiotracers. Consequently, this allows the radiochemist to implement a more complex chemical synthesis towards the imaging agent. PET imaging is utilised to obtain information on the non-invasive observation of chemical processes in tissues/organs. The radiotracer locates and binds to the receptor of interest and it is important that the imaging agent does not lead to significant receptor occupation [298].
Molecular imaging technology is not exploited to its full potential because many 18F-labelled probes are inaccessible or notoriously difficult to produce. In certain situations, the radiochemist faces synthesis challenges associated with the short half-life of 18F. Other issues are the use of sub-stoichiometric amounts of 18F compared to the precursor used and the availability of parent 18F sources [299].
Researchers are developing methods to access [18F]CF3-substituted molecules for the application in pharmaceutical discovery programmes (Figure 86).
![PET imaging agents [18F]fluxetin and [18F]flutamide](https://openmedscience.com/wp-content/uploads/2017/05/Figure-86-mm.jpg)
Figure 86. PET imaging agents [18F]fluxetin and [18F]flutamide.
Huiban et al. have reported the development of a process for the late-stage [18F]trifluoromethylation of (hetero)arenes from [18F]fluoride using commercially available reagents and (hetero)aryl iodides. This [18F]CuCF3 based protocol benefits from a large substrate scope and is characterised by its operational simplicity [300].
The introduction of fluorine or a trifluoromethyl group can improve the effects of medicinal drugs due to its stereoelectronic effects. Consequently, about 25% of all commercial drugs contain fluorine [301]. This analysis demonstrates how important it is to develop new synthesis methods to incorporate fluorine-18 into drug molecules. Numerous methods for fluorine incorporation are inadequate due to reaction step times and purification methods [302].
The research group of Yamamoto has developed a range of carbo- and heterocyclic molecules containing the trifluoromethyl group from a single starting material [303]. Central to this synthesis was the transformation of squaric acid to incorporate the CF3 group to generate various cyclic building blocks (Figure 87).
The squaric acid approach was attractive due to its four-carbon cyclic ring strain. The ring strain is released to form five and six membered cyclic molecules. The trifluoromethyl group was successfully introduced into the squarate in two steps in a single flask using the Ruppert-Prakash reagent [304]. The trifluoromethyl present in the squarate derivative can undergo rearrangement to various 1,4-benzoquinones which are present in many biologically significant molecules such as ubiquinone, vitamin K, antibiotic and anticancer agents [304].
This method generates various trifluoromethyl-containing polycyclic quinones in a few steps involving thermal ring expansion and aerobic oxidation which would otherwise be a difficult synthesis. These building blocks are capable of transforming to other complexed heterocyclic compounds such as butenolides and bicyclic pyridones including a five-carbon cyclic compound amino cyclopentenedione.
This methodology can be used to incorporate the trifluoromethyl group to create many interesting imaging agents.
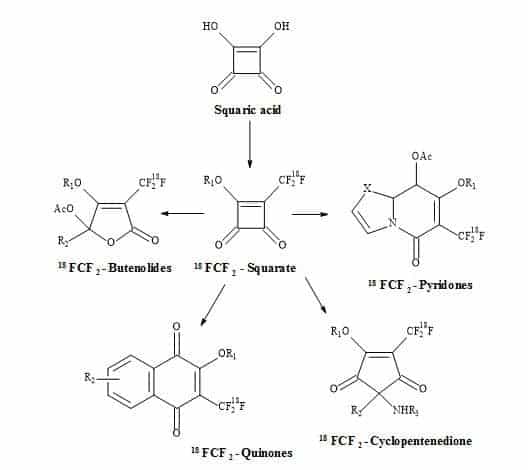
Figure 87. [18F]Squarate leading to highly functionalized [18F]compounds.
Zirconium-89 is a positron-emitting radionuclide used in PET imaging for the detection of oncology disease states [305]. This radiometal has a longer half-life of 79.3 hours compared to fluorine-18. This physical property makes it a more attractive radionuclide for the synthesis of PET radiotracers [306]. Zirconium-89 can bind to the desferrioxamine (DFO) ligand. DFO is a bacterial siderophore utilised since the late 1960s to treat iron overload in the human body [307].
The three hydroxamic acid moieties in DFO can form coordination bonds with Fe3+ ions; moreover, making DFO a hexadentate ligand that chelates the Fe3+ ions. Due to the coordination geometry of zirconium-89, DFO has also been used as a chelator in the design of PET imaging agents [308].
The work of Donnelly et al. producing designer novel DFO ligands containing squaryl functionality (Figure 88) [309]. The zirconium-89 is fixed by chelation to from the DFO. Consequently, the terminal squaric monoamide monoester can conjugate with biomolecules (e.g. herceptin (trastuzumab), rituximab and cetuximab) through the formation of a squaramide linker [28].
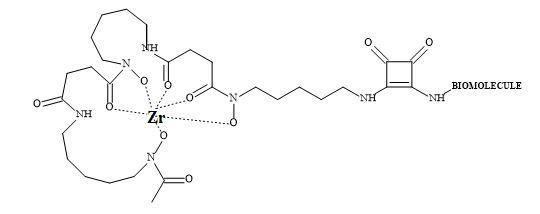
Figure 88. Zirconium-89 DFO-squaramide PET imaging agent.
Other radionuclides can be used such as gallium-68 for PET imaging [310]. In addition to indium-111 for SPECT imaging [311] to produce specific imaging agents to target tumours [312].
Magnetic resonance imaging
Magnetic resonance imaging (MRI) is a clinical tool for anatomical and functional imaging of diseased soft tissues, including solid tumours. MRI contrast agents can be used on patients to help in the detection of tumours. Gadolinium-based contrast agents are the most commonly used in clinical MRI. The challenge is to design and develop novel Gd(III) contrast agents with high relaxivity, low toxicity, and specific tumour binding properties [313].
Chemical modification to MRI contrast agents containing Gd(III) can increase their relaxation properties. The toxicity of Gd(III) contrast agents can be controlled by increasing the thermodynamic and kinetic stability profiles. These modifications contribute to an improved pharmacokinetic profile. The knowledge gained from cancer genomics and biology can provide vital information in designing tumour specific contrast agents [314].
Figure 89. Squaramide contrast agent for MRI.
Numerous Gd(III) chelates are investigated in animal models with the aim to provide superior MRI agents for oncology diseases. For example, the Gd(III) complex of inulin-1,4,7,10-tetraazacyclododecan-1,4-7-triacetic acid conjugate linked with squaramide as a contrast agent for MRI (Figure 89) [315].
Future challenges
The 3-D model of HIV reveals the active site is at the end of a cylindrical opening where the two aspartic acid residues lie. The diameter of this opening is the same as a C-60 Buckminsterfullerene. The group of Kenyon has been looking at how to utilise this fact by incorporating functional groups on the outer surface of the C-60 molecule [316]. They have managed to create a diamine C-60 derivative to be used in the research of protease inhibitors (Figure 90). An extension of this concept is to introduce squarate entities such as nitronium squarate [317] that can behave as molecular scissors. With the advent of nanotechnology, these types of chemical scissors may be advantageous in drug design [318].
Figure 90. C60 –Squaramide molecular scissors.
Combinatorial chemistry and vast drug substrate libraries can be produced simultaneously and quickly by automated procedures [319]. These compound libraries are a useful resource for the pharmaceutical industry in their quest for biologically active lead structures. The introduction of chloromethylated polystyrene support on one of the amino groups in squaramide provides a platform for chemical synthesis [24]. This squaryl resin is capable of reacting with a host of alkyl or aromatic alkyl aldehydes to produce secondary amino squarate supported substrates. The squaryl family provides suitable resin templates that can undergo screening for biological activity.
Figure 91. Squaramide DNA.
Cyclic peptides and their derivatives are often more stable in vivo than their linear counterparts and therefore can often represent promising drug candidates [320]. Peptide nucleic acids are a combination of amino acids linked to nucleobases. These peptide units are stable to snake venom phosphodiesterase and the squaryl equivalents may offer a broad range of drugs. The nucleoside and squaryl linkages should also be stable to nucleolytic cleavage [321,322]. These concepts can extend to molecular recognition patterns in the creation of squaramide DNA (Figure 91).
Acknowledgement
The author would like to thank Olivera Kitson for proofreading and making valuable changes to this review.
Conflict of interest
The author confirms that this article content has no conflict of interest.
References
Key References: 9, 10, 13, 17, 51, 52, 53, 54, 98, 100, 113, 130, 131, 165, 176, 182, 303
- Langmuir I. Isomorphism, isosterism and covalence. J Am Chem Soc. 1919; 41: 1543-1559. CrossRef
- Grimm HG. Structure and size of the non-metallic hydrides. Z. Electrochem. 1925; 31: 474-480.Reference Source
- Grimm HG. On the systematic arrangement of chemical compounds from the perspective of research on atomic composition; and on some challenges in experimental chemistry. Naturwissenschaften. 1929; 17: 557-564.Reference Source
- Erlenmeyer H, Leo M. On pseudoatoms. Helv Chim Acta.1932; 15: 1171-1186.Reference Source
- Friedman HL. Influence of isosteric replacements upon biological activity, Washington, EUA, National Academy of Science. 1951; 206: 295.Reference Source
- Thornber CW. Isosterism and molecular modification in drug design. Chem Soc Rev. 1979; 8: 563-580.CrossRef
- Burger A. Medicinal Chemistry, 3rd Ed., NY, EUA, Wiley 1970: 127.Reference Source
- Nicholls A, McGaughey GB, Sheridan RP, et al. Molecular shape and medicinal chemistry: a perspective. J Med Chem. 2010; 53(10): 3862-3886.CrossRef PubMed
- Patani GA, LaVoie EJ. Bioisosterism: A rational approach in drug design. Chem Rev. 1996; 96(8): 3147-3176.Reference Source
- Ballatore C, Huryn DM, Smith AB. Carboxylic acid (bio)isosteres in drug design. Chem Med Chem. 2013; 8(3): 385-395.CrossRef PubMed
- Brück A, McCoy LL, Kilway KV. Hydrogen bonds in carboxylic acid-carboxylate systems in solution. 1. In anhydrous, aprotic media. Org Lett. 2000; 2(14): 2007-2009.Reference Source
- Huggins DJ, Sherman W, Tidor B. Rational approaches to improving selectivity in drug design. J Med Chem. 2012; 55(4): 1424-1444.CrossRef PubMed
- Lima LM, Barreiro EJ. Bioisosterism: a useful strategy for molecular modification and drug design. Curr Med Chem. 2005; 12: 23-49.CrossRef PubMed
- Lounnasa V, Ritschelb T, Kelderc J, McGuired R, Bywatere RP, Foloppef N. Current process in structure-based rational drug design marks a new mindset in drug discovery. Computat Struct Biotechnol J. 2013; 5(6): 1-14.Reference Source
- Ooms F. Molecular modeling and computer aided drug design. Examples of their applications in medicinal chemistry. Curr Med Chem. 2000; 7(2): 141-158.CrossRef PubMed
- Macchiarulo A, Pellicciari R. Exploring the other side of biologically relevant chemical space: Insights into carboxylic, sulfonic and phosphonic acid bioisosteric relationships. J Mol Graph Model. 2007; 26(4): 728-739.CrossRef PubMed
- Charton J, Charruault L, Deprez-Poulain R, Deprez B. Alkylsquarates as a key intermediates for the rapid preparation of original drug-inspired compounds. Comb Chem High T Scr. 2008; 11(4): 294-303.CrossRef
- Corey EJ. The logic of chemical synthesis: multistep synthesis of complex carbogenic molecules (Nobel Lecture). Angew Chem Int Edn Engl. 1991; 30: 455-465.CrossRef
- Tietgen H, Walden M. In Drug Discovery and Evaluation: Safety and Pharmacokinetic Assays; H. Gerhard Vogel, Jochen Maas, Franz J. Hock, Dieter Mayer, E.ds., Springer Berlin Heidelberg, Physicochemical Properties, 2013, pp 1125-1138.Reference Source
- Lipinski CA, Lombardo F, Dominy BW, Feeney P. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. J Adv Drug Delivery Rev. 1997; 23: 3-25.CrossRef
- Pople JA, Gordon M. Molecular orbital theory of the electronic structure of organic compounds. I. Substituent effects and dipole moments. J Am Chem Soc. 1967; 89 (17): 4253-4261.CrossRef
- Kubinyi H. (2001) Hydrogen Bonding: The Last Mystery in Drug Design? In Pharmacokinetic Optimization in Drug Research: Biological, Physicochemical, and Computational Strategies (eds B. Testa, H. van de Waterbeemd, G. Folkers and R. Guy), Verlag Helvetica Chimica Acta, Zürich.Reference Source
- Williams, Wilkins Lippincott. In Foye’s Principles of Medicinal Chemistry; Thomas L. Lemke, David A. Williams, Victoria F. Roche, S. William Zito. Eds.,Lippincott Williams & Wilkins, 2013.Reference Source
- Maahs G, Hegenberg P. Syntheses and derivatives of squaric acid. Angew Chem Int Ed. Engl. 1966; 5(10): 888-893.CrossRef
- Storer RI, Aciroa C, Lyn H, Jones LH. Squaramides: physical properties, synthesis and applications. Chem Soc Rev. 2011; 40: 2330-2346.CrossRef
- Lim NC, Morton MD, Jenkins HA, Brückner C. Squaric acid N-hydroxylamides: synthesis, structure, and properties of vinylogous hydroxamic acid analogues. J Org Chem. 2003; 68(24): 9233-9241.Reference Source
- Lassalas P, Gay B, Lasfargeas C, et al. Structure property relationships of carboxylic acid isosteres. J Med Chem. 2016; 59: 3183-3203.CrossRef
- Wurm FR, Klok H-A. Review article be squared: expanding the horizon of squaric acid-mediated conjugations. Chem Soc Rev. 2013; 42: 8220-8236.Reference Source
- Ishida T, Shinada T, Ohfune Y. Synthesis of novel amino squaric acids via addition of dianion enolates derived from N-Boc amino acid esters. Tetrahedron Lett. 2005; 46(2): 311-314.CrossRef
- Onaran MB, Comeau AB, Seto CT. Squaric acid-based peptidic inhibitors of matrix metalloprotease-1 (MMP-1). J Org Chem. 2005; 70(26): 10792–10802.PubMed
- Lim NC, Morton MD, Jenkins HA, Bruckner C. Squaric acid N-hydroxylamides: synthesis, structure, and properties of vinylogous hydroxamic acid analogues. J Org Chem. 2003; 68: 9233-9241.CrossRef
- van Rossum JM. The relation between chemical structure and biological activity. J Pharm Pharmacol. 1963; 15: 285–316.Reference Source
- Manallack DT, Prankerd RJ, Yuriev E, Oprea TI, Chalmers DK. The significance of acid/base properties in drug discovery. Chem Soc rev. 2013; 42(2): 485-496.CrossRef PubMed
- Day RO, McLachlan AJ, Graham GG, Williams KM. Pharmacokinetics of nonsteroidal anti-inflammatory drugs in synovial fluid. Clin Pharmacokinet. 1999; 36(3): 191-210.CrossRef PubMed
- Wagner JR, Lee CT, Durrant JD, Malmstrom RD, Feher VA, Amaro RE. Emerging computational methods for the rational discovery of allosteric drugs. Chem Rev. 2016; 116(11):6370-6390.CrossRef PubMed
- Mani CM, Berthold T, Fechler N. “Cubism” on the nanoscale: from squaric acid to porous carbon cubes. Small. 2016; 12(21): 2906-2912.CrossRef PubMed
- Gu L, Mooney DJ. Biomaterials and emerging anticancer therapeutics: engineering the microenvironment. Nat Rev Cancer. 2016; 16, 56-66.CrossRef PubMed
- Cook D, Brown D, Alexander R, March R, et al. OUTLOOK: Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. 2014; 13: 419-431.PubMed
- Sethy SP, Meher CP, Biswal S, Sahoo U, Patro SK. The role of bioisosterism in molecular modification and drug design: a review. AJPSR. 2013; 3(1): 61-87.Reference Source
- Meanwell NA. Synopsis of some recent tactical application of bioisosteres in drug design. J Med Chem. 2011; 54(8):2529-2591.CrossRef PubMed
- Cheeseright T. The identification of bioisosteres as drug development candidates. Innovations in Pharmaceutical Technology. 2009; 28: 22-26.Reference Source
- Brown N. (2012) Bioisosterism in Medicinal Chemistry, in Bioisosteres in Medicinal Chemistry (ed N. Brown), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany.Reference Source
- Pharmacokinetics and Metabolism in Drug Design Edited by D. A. Smith, H. van de Waterbeemd, D. K. Walker, R. Mannhold, H. Kubinyi, H. Timmerman Wiley-VCH Verlag GmbH, 2001.Reference Source
- Mandal S, Moudgil M, Mandal SK. Rational drug design. Eur J Pharmacol. 2009; 625: 90-100.CrossRef
- Camenischa G, Folkersb G, van de Waterbeem H. Review of theoretical passive drug absorption models: historical background, recent developments and limitations. Pharma Acta Helv. 1996; 71(5): 309-327.CrossRef PubMed
- Lipophilicity in Drug Action and Toxicology, 1996 VCH Verlagsgesellschaft mbH, Editor(s): Vladimir Pliška, Bernard Testa, Han van de Waterbeemd.Reference Source
- Mavromoustakos T, Durdagi S, Koukoulitsa C, et al. Strategies in the rational drug design. Curr Med Chem. 2011; 18(17): 2517-2530.CrossRef PubMed
- Pinter T, Jana S, Courtemanche RJM, Hof F. Recognition properties of carboxylic acid bioisosteres: anion binding by tetrazoles, aryl sulfonamides, and acyl sulfonamides on a calix[4]arene scaffold. J Org Chem. 2011; 76(10): 733-3741.CrossRef PubMed
- Ali G, Subhan F, Islam N, et al. Input of isosteric and bioisosteric approach in drug design. J Chem Soc Pak. 2014; 36(1).Reference Source
- Gong-Xin He, Jeffrey P, Krise RO. Prodrugs Biotechnology: Pharmaceutical Aspects Volume V, 2007, pp 923-964. Prodrugs of Phosphonates, Phosphinates, and Phosphates.Reference Source
- Holt DA, Levy MA, Yen H-Y, Oh H-J, Metcalf BW, Wier PJ. Inhibition of steroid 5α-reductase by 3-nitrosteroids: synthesis, mechanism of inhibition, and in vivo activity. Bioorg Med Chem Lett. 1991; 1(1): 27-32.Reference Source
- Holt DA, Levy MA, Ladd DL, et al. Steroidal A ring aryl carboxylic acids: a new class of steroid 5.alpha.-reductase inhibitors. J Med Chem. 1990; 33(3): 937-942.CrossRef
- Ueda T, Crast B, Mikkilineni AB, Partyka RA. Synthesis of phenoxyacetyl-N-(hydroxydioxocyclobutenyl)cycloserines. Tetrahedron Lett. 1991; 32(31): 3767-3770.CrossRef
- Soll RM, Kinneya WA. Primeaua J, et al. 3-Hydroxy-3-cyclobutene-1,2-dione: application of novel carboxylic acid bioisostere to an in-vivo active non-tetrazole angiotensin-II antagonist. Bioorg Med Chem Lett. 1993; 3(4):757-760.CrossRef
- Pirrung MC, Han H, Ludwig RT. Inhibitors of thermus thermophilus isopropylmalate dehydrogenase. J Org Chem. 1994; 59(9): 2430-2436.CrossRef
- West R, Powell DL. New aromatic anions. III. Molecular orbital calculations on oxygenated anions. J Am Chem Soc. 1963; 85(17): 2577-2579.CrossRef
- Allen FH, Cruz-Cabeza AJ, Wood PA, Bardwell DA. Hydrogen bond landscapes, geometry and energetics of squaric acid and its mono- and dianions: a Cambridge structural database, IsoStar and computational study. Acta Cryst. 2013; B69: 514-523.CrossRef PubMed
- Bao X, Zhou X, Lovitt CF, et al. Molecular Orbitals of the oxocarbons (CO)n, n = 2–6. Why does (CO)4 have a triplet ground state? J Am Chem Soc. 2012; 134(24), 10259-1027.PubMed
- de Oliveira, V. E., Diniz, R., Machado, F. C. and de Oliveira, L. F. C. (2016) Oxocarbons, Pseudo-oxocarbons, and Squaraines, in Cross Conjugation: Modern Dendralene, Radialene and Fulvene Chemistry (eds H. Hopf and M. S. Sherburn), Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany.Reference Source
- Seitz G, Imming P. Oxocarbons and pseudooxocarbons. Chem Rev. 1992; 92(6): 1227-1260.CrossRef
- von Ragué Schleyer P, Najafian K, Kiran B, Jiao H. Are oxocarbon dianions aromatic? J Org Chem. 2000; 65 (2): 426-431.Reference Source
- Eggerding D, West R. Synthesis of dihydroxycyclopropenone (deltic acid). J Am Chem Soc. 1975; 97 (1): 207-208.CrossRef
- Lautié A, Lautié MF, Novak A. Vibrational spectra and valence force field calculation of deltic acid. J Mol Struct. 1986; 142: 29-32.CrossRef
- Eggerding D, West R. Synthesis and properties of deltic acid (dihydroxycyclopropenone) and the deltate ion. J Am Chem Soc. 1976; 98 (12): 3641-3644.CrossRef
- West R, Niu H-Y, Powell DL, Evans MV. Symmetrical resonance stabilized anions, 1 CnOn-2. J Am Chem Soc. 1960; 82 (23): 6204-6205.Reference Source
- Ireland DT, Walton HF. Ionization of diketocyclobutenediol and its metal complexes. J Phys Chem. 1967; 71(3): 751-754CrossRef
- Tedesco PH, Walton HF. Metal complexes of squaric acid (diketocyclobutenediol) in aqueous solution. Inorg Chem. 1969; 8(4): 932-937.CrossRef
- MacDonald DJ. Ionization constants of squaric acid. J Org Chem. 1968; 33(12): 4559-4560.CrossRef
- Park JD, Cohan S, Lacher JR. Hydrolysis Reactions of Halogenated Cyclobutene Ethers: Synthesis of Diketocyclobutenediol. J Am Chem Soc. 1962; 84: 2919.CrossRef
- Alexandersson D, Vannerberg N-G. The stability constants of squaric acid and rhodizonic acid. Acta Chem Scand. 1972; 5: 1909-1920.Reference Source
- Gelb RI, Schwartz LM, Laufer DA. Structure of moderately strong acids in aqueous solution. J Am Chem Soc. 1981; 103 (19): 5664-5673.CrossRef
- Perez GV, Perez AL. Organic acids without a carboxylic acid functional group. J Chem Education. 2000; 77(7): 910-915.CrossRef
- Wang Y, Stucky GD, Williams JM. Is squaric acid square? A combined X-ray and neutron diffraction study of 3,4-dihydroxycyclobut-3-ene-1,2-dione. J Chem Soc Perkin Trans 2. 1974; 35-38.CrossRef
- Tempest PA, Armstrong RW. Cyclobutenedione derivatives on solid support: towards multiple core structure libraries. J Am Chem Soc. 1997; 119: 7607-7608.CrossRef
- Lui H, Tomooka CS, Moore HW. An efficient general synthesis of squarate esters. Synthetic Commun. 1997; 27(12): 2177-2180.Reference Source
- Cohen S, Cohen S. Preparation and reactions of derivatives of squaric acid, alkoxy-, hydroxyl, and aminocyclobutenediones. J Am Chem Soc. 1966; 88(7): 1533-1536.Reference Source
- Schmidt AH, Ried W. Die preparative chemie der cyclobutendione: 111. Synthese von quadratsäure, benzocyclobutendion und deren derivaten. Synthesis. 1978: 869-880.Reference Source
- Paquette LA. Cascade rearrangements following twofold addition of alkenyl anions to squarate esters. Eur J Org Chem. 1998: 1709-1728.CrossRef
- Ohno M, Yamamoto Y, Shirasaki Y, Eguchi S. Synthesis of squaric acid derivatives by lewis acid-catalysed reaction of its dichloride, methyl ester chloride, diethylamide chloride, and ethyl diester with unsaturated organosilanes: new method for C-C bond formation on cyclobutenedione. J Chem Soc Perkin Trans 1. 1993: 263-271.CrossRef
- Yamamoto Y, Ohno M, Eguchi S. Ring Transformation of 4-acylmethyl-2-chloro-4-hyroxy-2-cyclobutenone to γ-acylmethylenetetronate by thermal rearrangement: new synthetic aspect of squaric acid as a C4-synthon. Tetrahedron. 1994; 50(26): 7783-7798.Reference Source
- Fu N, Allen AD, Kobayashi S, Tidwell TT, Vukovic S. Structural effects on interconversion of oxygen-substituted biketenes and cyclobutenediones. J Org Chem. 2008; 73: 1768-1773.Reference Source
- Li B-S, Wang Y, Jin Z, Zheng P, Ganguly R, Chi YB. Carbon-carbon bond activation of cyclobutenones enabled by the addition of chiral organocatalyst to ketone. Nat Commun. 2015; 6: 6207.PubMed
- Yerxa BR, Yang K, Moore HW. Synthesis of (±)-septicine. Tetrahedron. 1994; 50(21): 6173-6180.CrossRef
- Enhsen A, Karabelas K, Heerding, JM, Moore HW. Synthesis of hydroxyquinones and related compounds: semisquaric acids, (±)-terreic acid, (±)-perezone and (±)-isoperezone. J Org Chem. 1990; 55 (4): 1177-1185.Reference Source
- Heerding JM, Moore HW. Regiospecific synthesis of hydroxyquinones and related compounds from 3-tert-butoxycyclobutene-1,2-dione. J Org Chem. 1991; 56(12): 4048-4050.CrossRef
- Casiraghi G, Zanardi F. The vinylogous aldol reaction: a valuable, yet understated carbon-carbon bond-forming maneuver. Chem Rev. 2000; 100(6): 1929-1972.Reference Source
- Ohno M, Yamamoto Y, Eguchi S. Squaric acid as synthon for highly selective synthesis of polyfunctionalized carbo- and heterocycles. J Syn Org Chem Jpn. 1997; 55(9): 785-792.Reference Source
- Azizi N, Davoudpour A, Eskandari F, Batebi E. Squaric acid catalyzed simple synthesis of N-substituted pyrroles in green reaction media. Monatshefte für Chemie – Chemical Monthly. 2013; 144(3): 405-409.CrossRef
- Peña-Cabrera E, Liebeskind LS. Squaric acid ester-based total synthesis of echinochrome A. J Org Chem. 2002; 67(5): 1689-1691.PubMed
- Konovalova A, Søgaard-Andersen L, Kroos L. Regulated proteolysis in bacterial development. FEMS Microbiol Rev. 2014; 38(3): 493-522.CrossRef PubMed
- Comins DL, Chen X, Morgan LA. Enantiopure N-acyldihydropyridones as synthetic intermediates: asymmetric synthesis of (−)-septicine and (−)-tylophorine. J Org Chem. 1997; 62 (21): 7435-7438.PubMed
- Burmeister HR, Ciegler A, Vesonder RF. Moniliformin, a metabolite of fusarium moniliforme NRRL 6322: purification and toxicity. Appl Environ Microb. 1979; 37(1): 11-13.PubMed
- von Bargen KW, Lohrey L, Cramer B, Humpf HU. Analysis of the fusarium mycotoxin moniliformin in cereal samples using 13C2-moniliformin and high-resolution mass spectrometry. J Agric Food Chem. 2012; 60(14): 3586-3591.Reference Source
- Rabie CJ, Marasas WF, Thiel PG, Lübben A, Vleggaar R. Moniliformin production and toxicity of different fusarium species from Southern Africa. Appl Environ Microb. 1982; 43(3): 517-521.PubMed
- Bräse S, Encinas A, Keck J, Nising CF. Chemistry and biology of mycotoxins and related fungal metabolites. Chem Rev. 2009; 109(9): 3903-3990.Reference Source
- Ohno M, Eguchi S. Directed Synthesis of biologically interesting heterocycles with squaric acid (3,4-dihydroxy-3-cyclobutene-1,2-dione) based technology. Top Heterocycl Chem. 2006; 6: 1-37.CrossRef
- Springer JP, Clardy J, Cole RJ, et al. Structure and synthesis of moniliformin, a novel cyclobutane microbial toxin. J Am Chem Soc. 1974; 96(7): 2267-2268.CrossRef PubMed
- Mukkanti A, Periasamy M. Methods of synthesis of cyclobutenediones. ARKIVOC. 2005; (xi): 48-77.Reference Source
- Shinada T, Isida T, Ohfune Y. Recent progress on squaric acid research in bioorganic fields. J Syn Org Chem Jpn. 2007; 65(1): 30-43.CrossRef
- Cohen S, Lacher JR, Park JD. Diketocyclobutenediol. J Am Chem Soc. 1959; 81(13): 3480-3480.CrossRef
- Heys JR, Chew EH. Synthesis of [14C4]squaric acid. J Label Compd Radiopharm. 1984; 21(5): 409-413.CrossRef
- Bellus D. Synthesis and reactivity of compounds with cyclobutane ring(s). 10. Syntheses of squaric acid, its monoorthoesters, and related derivatives via [2+2] cycloadditions of tetraalkoxyethylenes with heterosubstituted ketenes. J Org Chem. 1979; 44(8): 1208-1211.CrossRef
- Chickos JS. Methylhydroxycyclobutenedione. J Am Chem Soc. 1970; 92 (19): 5749-5750.CrossRef
- Kraus JL. Reactivity of organo lithium reagents on dimethyl squarate: a 1,2-addition process leading to new 2-hydroxy-3,4-dimethoxy-3-cyclobutenone. Tetrahedron Lett. 1985; 26(15): 1867-1870.CrossRef
- Dehmlow EV, Schell HG. Darstellung von 3-alkoxy-4-alkyl-3-cyclobuten-1,2-dionen. Chem. Ber. 1980; 113: 1-8.CrossRef
- Liebeskind LS, Fengl RW, Wirtz KR, Shawe TT. An improved method for the synthesis of substituted cyclobutenediones. J Org Chem. 1988; 53(11): 2482-2488.CrossRef
- Reed MW, Pollart DJ, Perri ST, Foland LD, Moore HW. Synthesis of 4-substituted-3-alkoxy-3-cyclobutene-1,2-diones. J Org Chem. 1988; 53(11): 2477-2482.CrossRef
- Dubois J, Bory S, Gaudry M, Marquet A. Vitamin K dependent carboxylation: synthesis and biological properties of tetrazolyl analogs of pentapeptidic substrates. J Med Chem. 1984; 27(9): 1230-1233.CrossRef
- Sosnovsky G, Lukszo J, Gravela E, Zuretti MF. In search of new anticancer drugs. 13. Phosphonic and phosphinic analogs of ornithine. J Med Chem. 1985; 28(9): 1350-1354.CrossRef
- Serratosa F. Acetylene diethers: a logical entry to oxocarbons. Acc Chem Res. 1983; 16(5): 170-176.CrossRef
- Pryde DC, Jones LH, Gervais DP, et al. Selection of a novel anti-nicotine vaccine: influence of antigen design on antibody function in mice. PLoS One. 2013; 8(10): e76557.CrossRef PubMed
- Butera JA, Antane MM, Antane SA, et al. Design and SAR of novel potassium channel openers targeted for urge urinary incontinence. 1. N-cyanoguanidine bioisosteres possessing in vivo bladder selectivity. J Med Chem. 2000; 43(6): 1187-1202.CrossRef PubMed
- Gilbert AM, Antane MM, Argentieri TM, et al. Design and SAR of novel potassium channel openers targeted for urge urinary incontinence. 2. Selective and potent benzylamino cyclobutenediones. J Med Chem. 2000; 43(6): 1203-1214.CrossRef PubMed
- West R, editor (1980), Oxocarbons. Academic Press, New York.Reference Source
- Triebs A, Jacob K. Cyclotrimethine dyes derived from squaric acid. Angew Chem Int Ed. 1965; 4:694.CrossRef
- Lynch DE. Pyrrolyl squaraines – fifty golden years. Metals. 2015; 5: 1349-1370.CrossRef
- Johnson RL, Koerner JF. Excitatory amino acid neurotransmission. J Med Chem. 1988; 31(11): 2057-2066.Reference Source
- Wagner I, Musso H. New naturally occurring amino acids. Angew. Chem Int Ed. Engl. 1983; 22: 816-828.CrossRef
- Kudo F, Miyanaga A, Eguchi T. Biosynthesis of natural products containing β-amino acids. Nat Prod Rep. 2014; 31(8): 1056-1073.CrossRef
- Petrenko AB, Yamakura T, Baba H, Shimoji K. The role of N-methyl-D-aspartate (NMDA) receptors in pain: A review. Anesth Analg. 2003; 97: 1108-1116.CrossRef
- Swope SL, Moss SJ, Raymond LA, Huganir RL. Regulation of ligand-gated ion channels by protein phosphorylation. Adv Second Messenger Phosphoprotein Res. 1999; 33: 49-78.CrossRef PubMed
- Lee C-H, Lü W, Michel JC, et al. NMDA receptor structures reveal subunit arrangement and pore architecture. Nature. 2014; 511: 191-197.CrossRef PubMed
- Newcomer JW, Farber NB, Olney JW. NMDA receptor function, memory, and brain aging. Dialogues Clin Neurosci. 2000; 2(3): 219-232.PubMed
- Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL): CRC Press/Taylor & Francis; 2009.Reference Source
- Zhou X, Hollern D, Liao J, Andrechek E, Wang H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013; 4: e560.CrossRef PubMed
- Dong X, Wang Y, Qin Z. Molecular mechanisms of excitotoxicity and their relevance to pathogenesis of neurodegenerative diseases. Acta Pharmacol Sin. 2009; 30(4): 379-387.CrossRef PubMed
- Murray PS, Holmes PV. An overview of brain-derived neurotrophic factor and implications for excitotoxic vulnerability in the hippocampus. Int J Pept. 2011; 2011: 654085.CrossRef PubMed
- Steeds H, Carhart-Harris RL, Stone JM. Drug models of schizophrenia. Ther Adv Psychopharmacol. 2015; 5(1): 43-58.CrossRef PubMed
- Monaghan DT, Jane DE. Pharmacology of NMDA receptors. In: Van Dongen AM, editor. Biology of the NMDA Receptor. Boca Raton (FL): CRC Press; 2009. Chapter 12.PubMed
- Kinney WA, Lee NE, Garrison DT, et al. Bioisosteric replacement of the alpha-amino carboxylic acid functionality in 2-amino-5-phosphonopentanoic acid yields unique 3,4-diamino-3-cyclobutene-1,2-dione containing NMDA antagonists. J Med Chem. 1992; 35(25): 4720-4726.CrossRef PubMed
- Kinney WA, Abou-Gharbia M, Garrison DT, et al. Design and synthesis of [2-(8,9-dioxo-2,6-diazabicyclo[5.2.0]non-1(7)-en-2-yl)-ethyl]phosphonic acid (EAA-090), a potent N-methyl-D-aspartate antagonist, via the use of 3-cyclobutene-1,2-dione as an achiral alpha-amino acid bioisostere. J Med Chem. 1998; 41(2): 236-246.PubMed
- Liebeskind LS, Fengl RW. 3-Stannylcyclobutenediones as nucleophilic cyclobutenedione equivalents. Synthesis of substituted cyclobutenediones and cyclobutenedione monoacetals and the beneficial effect of catalytic copper iodide on the Stille reaction. J Org Chem. 1990; 55 (19): 5359-5364.CrossRef
- Cordovilla C, Bartolomé C, Martínez-Ilarduya JM, Espinet P. The Stille reaction, 38 Years Later. ACS Catalysis. 2015; 5(5): 3040-3053.CrossRef
- Dhanoa DS, Bagley SW, Chang RSL, et al. Non-peptide angiotensin II receptor antagonists. Design, synthesis, and biological activity of N-substituted indoles and dihydroindoles. J Med Chem. 1993; 36(26): 4230-4238.CrossRef
- Noda K, Saad Y, Kinoshita A, et al. Tetrazole and carboxylate groups of angiotensin receptor antagonists bind to the same subsite by different mechanisms. J Biol Chem. 1995; 270(5): 2284-2289.Reference Source
- Soll RM, Kinneya WA, Primeaua, J, et al. 3-Hydroxy-3-cyclobutene-1,2-dione: Application of novel carboxylic acid bioisostere to an in-vivo active non-tetrazole angiotensin-II antagonist. Bioorg Med Chem Lett. 1993, 3(4), 757-760.Reference Source
- Ong CKS, Lirk P, Tan CH, Seymour RA. An evidence-based update on nonsteroidal anti-inflammatory drugs. Clin Med Res. 2007; 5(1): 19-34.CrossRef PubMed
- Petersen S, Müller E. Über eine neue Gruppe von Süßstoffen. Chem Ber. 1948; 81: 31-38.Reference Source
- Muller GW, Culberson JC, Roy G, et al. Carboxylic acid replacement structure-activity relationships in suosan type sweeteners. A sweet taste antagonist. J Med Chem. 1992; 35(10): 1747-1751.CrossRef
- Tinti J-M, Nofre C. Design of sweeteners. Sweeteners. 1991: 88-99.CrossRef
- Nozaki Y, Katayama N, Ono H, et al. Binding of a non-β-lactam antibiotic to penicillin-binding proteins. Nature. 1987; 325: 179-180.CrossRef
- Stammer CH, Kartha CK, Chaturvedi NC, McKinney JD. Cycloserine derivatives. J Med Chem. 1970; 13(5): 1013-1015.CrossRef
- Bernstein KE, Ong FS, Blackwell W-L B, et al. A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol Rev. 2013; 65: 1-46.CrossRef PubMed
- Song JC, White CM. Clinical pharmacokinetics and selective pharmacodynamics of new angiotensin converting enzyme inhibitors: an update. Clin Pharmacokinet. 2002; 41(3): 207-224.CrossRef PubMed
- Lima DP de. Synthesis of angiotensin-converting enzyme (ACE) inhibitors: an important class of antihypertensive drugs ACE inhibitors are one of the most active classes of molecules that lower blood pressure. Química Nova. 1999; 22(3): 375-381.Reference Source
- Palomo C, Ganboa I, Oiarbide M, Sciano GT, Miranda JI. A β-lactam route to short peptide segments related to angiotensin converting enzyme (ACE) inhibitors. ARKIVOC. 2002; (v): 8-16.Reference Source
- Bartroli J, Ramis J, Marin A, Forn J. Synthesis of 3H-labelled enalapril maleate. J Label Compd Radiopharm. 1986; 23: 771-776.CrossRef
- Vagner J, Qu H, Hruby VJ. Peptidomimetics, a synthetic tool of drug discovery. Curr Opin Chem Biol. 2008; 2(3): 292-296.CrossRef PubMed
- Avan I, Hallb CD, Katritzky AR. Peptidomimetics via modifications of amino acids and peptide bonds. Chem Soc Rev. 2014; 43: 3575-3594.CrossRef PubMed
- Cosic I, Pirogova E. Bioactive peptide design using the resonant recognition model. Nonlinear Biomed Phys. 2007: 1:7.CrossRef PubMed
- Trabocchi A, Guarna A. Peptidomimetics in Organic and Medicinal Chemistry: The Art of Transforming Peptides in Drugs. 2014 John Wiley & Sons.Reference Source
- Martínez L, Sampedro A, Sanna, E, Costa A, Rotger C. Synthesis and conformational studies of peptido-squaramide foldable modules: a new class of turn-mimetic compounds. Org Biomol Chem. 2012; 10(9): 1914-1921.CrossRef
- Hanson GJ, Lindberg T. Synthesis of new dipeptide analogs containing novel ketovinyl and hydroxyethylidene isosteres via Grignard addition to chiral alpha-amino aldehydes. J Org Chem. 1985; 50(25): 5399-5401.CrossRef
- Ryan JW, Chung A, Ryan US. Angiotensin-converting enzyme: I. New strategies for assay. Environmental Health Perspectives. 1980; 35: 165-169.CrossRef
- Fisher NDL, Hollenberg NK. Renin Inhibition: What are the therapeutic opportunities? J Am Soc Nephrol. 2005; 16: 592-599.Reference Source
- Zambrowicz A, Timmer M, Polanowski A, Lubec, G, Trziszka T. Manufacturing of peptides exhibiting biological activity. Amino Acids. 2013; 44(2): 315-20.CrossRef PubMed
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today. 2015; 20(1): 122-128.CrossRef PubMed
- Liebeskind LS, Yu MS, Yu RH, Wang J, Hagen KS. 4,4′-Bi(cyclobutene-1,2-diones): bisquaryls. J Am Chem Soc. 1993; 115(20): 9048-9055.CrossRef
- Geng H, Jiang F, Wu Y-D. Accurate structure prediction and conformational analysis of cyclic peptides with residue-specific force fields. J Phys Chem Lett. 2016; 7(10): 1805-1810.CrossRef PubMed
- WHO. World Malaria Report 2015. Geneva, Switzerland: World Health Organization; 2015.Reference Source
- Travassos MA, Laufer MK. Resistance to antimalarial drugs: molecular, pharmacological and clinical considerations. Pediatr Res. 2009; 65(5 Pt 2): 64R-70R.CrossRef PubMed
- Kumar SP, Gloria PMC, Goncalves LM, et al. Squaric acid: a valuable scaffold for developing antimalarials? Med Chem Comm. 2012; 3: 489-493.CrossRef
- Gardner MJ, Hall N, Fung E, et al. Genome sequence of the human malaria parasite plasmodium falciparum. Nature. 2002; 419(6906): 498-511.CrossRef PubMed
- Marco M, Coterón JM. Falcipain inhibition as a promising antimalarial target. Curr Top Med Chem. 2012;12(5):408-44.CrossRef PubMed
- Malerich JP, Hagihara K, Rawal VH. Chiral squaramide derivatives are excellent hydrogen bond donor catalysts. J Am Chem Soc. 2008; 130 (44), 14416-14417.CrossRef PubMed
- Held FE, Tsogoeva SB. Asymmetric cycloaddition reactions catalyzed by bifunctional thiourea and squaramide organocatalysts: recent advances. Catal Sci Technol. 2016; 6: 645-667.CrossRef
- Varga E, Mika LT, Csámpai A, Holczbauer T, Kardosa G, Soós T. Mechanistic investigations of a bifunctional squaramide organocatalyst in asymmetric Michael reaction and observation of stereoselective retro-Michael reaction. RSC Adv. 2015; 5: 95079-95086.CrossRef
- Quiñonero D, Frontera A, Ballester P, Deya PM. A theoretical study of aromaticity in squaramide and oxocarbons. Tetrahedron Letts. 2000; 41(12): 2001-2005.Reference Source
- Robert B.P. Elmes & Katrina A. Jolliffe. Amino acid-based squaramides for anion recognition. Supramolecular Chemistry. 2015; 27(5-6): 321-328.Reference Source
- Kitson SL, Quinn DJ, Moody TS, Speed D, Watters W, Rozzell D. Antibody-drug conjugates (ADCs) – A new generation of biotherapeutic bullets. Chim Oggi. 2013; 31(4): 30-36.
- Kitson SL, Cuccurullo V, Moody TS, Mansi L. Radionuclide antibody-conjugates, a targeted therapy towards cancer. Curr Radiopharm. 2013; 6(2): 57-71.CrossRef PubMed
- Martin B, Lai X, Baettig U, et al. Early process development of a squaramide-based CXCR2 receptor antagonist. Org Process Res Dev. 2015; 19: 1038-1043.CrossRef
- Merritt RJ, Rokosza LL, Nelson KH, et al. Synthesis and structure–activity relationships of 3,4-diaminocyclobut-3-ene-1,2-dione CXCR2 antagonists. Bioorg Med Chem Lett. 2006; 16(15): 4107-4110.PubMed
- Chao J, Taveras AG, Chao J, et al. C(4)-alkyl substituted furanyl cyclobutenediones as potent, orally bioavailable CXCR2 and CXCR1 receptor antagonists. Bioorg Med Chem Lett. 2007; 17(13): 3778-3783.CrossRef PubMed
- Quintana M, Alegre-Requena JV, Marques-Lopez E, Herrera RP, Triola G. Squaramides with cytotoxic activity against human gastric carcinoma cells HGC-27: synthesis and mechanism of action. Med Chem Commun. 2016; 7: 550-561.CrossRef
- Sun L, Chiu D, Kowal D, et al. Characterization of two novel N-methyl-D-aspartate antagonists: EAA-090 (2-[8,9-Dioxo-2,6-diazabicyclo [5.2.0]non-1(7)-en2-yl]ethylphosphonic Acid) and EAB-318 (R-α-Amino-5-chloro-1-(phosphonomethyl)-1H-benzimidazole-2-propanoic acid hydrochloride). J Pharmacol ExpTher. 2004; 310(2): 563-570.CrossRef PubMed
- Pérez-Pinzón MA, Steinberg GK. CGS 19755 (Selfotel): a novel neuroprotective agent against CNS injury. CNS Drug Rev. 1996; 2(3): 257-268.PubMed
- Murphy DE, Hutchison AJ, Hurt SD, Williams M, Sills MA. Characterization of the binding of [3H]-CGS 19755: a novel N-methyl-D-aspartate antagonist with nanomolar affinity in rat brain. Br J Pharmacol. 1988; 95(3): 932-938.CrossRef PubMed
- Lee CW, Cao H, Ichiyama K, Rana TM. Design and synthesis of a novel peptidomimetic inhibitor of HIV-1 Tat-TAR interactions: squaryldiamide as a new potential bioisostere of unsubstituted guanidine. Bioorg Med Chem Lett. 2005; 15: 4243-4246.CrossRef PubMed
- Fu J, Xia A, Dai Y, Qi X. Design-Based peptidomimetic ligand discovery to target HIV TAR RNA using comparative analysis of different docking methods. Curr HIV Res. 2016 Jul 19. [Epub ahead of print].CrossRef PubMed
- MacLachlan LS, Rovner ES. New treatments for incontinence. Adv Chronic Kidney Dis. 2015; 22(4): 279-288.CrossRef PubMed
- Elzayat E, Khaled S, Kashiwabara T, Elhilali M, Corcos J. Effect of the potassium channel opener WAY-133537 on the overactive bladder of spinalized rats. Neurourol Urodyn. 2006; 25(7): 808-814.CrossRef PubMed
- Coghlan MJ, Carroll WA, Gopalakrishnan M. Recent developments in the biology and medicinal chemistry of potassium channel modulators: update from a decade of progress. J Med Chem. 200; 44 (11): 1627-1653.PubMed
- Porter JR, Archibald SC, Childs K, et al. Squaric acid derivatives as VLA-4 integrin antagonists. Bioorg Med Chem Lett. 2002; 12(7):1051-1054.CrossRef PubMed
- Kaku S, Isobe Y, Kiuchi Y, Tanaka M, Muramatsu M, Higuchi S. Interaction of the new histamine H2-receptor antagonist pibutidine hydrochloride with canine cloned H2-receptor expressed cells. Arzneimittelforschung. 1999; 49(1): 67-71.CrossRef PubMed
- Ventola CL. The antibiotic resistance crisis: part 1: causes and threats. Pharmacy and Therapeutics. 2015;40(4):277-283.PubMed
- Pokrovskayaa V, Baasov T. Dual-acting hybrid antibiotics: a promising strategy to combat bacterial resistance. Expert Opin Drug Discov. 2010; 5(9): 883-902.CrossRef PubMed
- Bremner JB, Kelso MJ. Synthesis of berberine–efflux pump inhibitor hybrid antibacterials. Synth commun. 2010; 40(23):3561-3568.PubMed
- O’Connell KMG, Hodgkinson JT, Sore HF, Welch M, Salmond GPC, Spring DR. Combating multidrug-resistant bacteria: current strategies for the discovery of novel antibacterials. Angew Chem Int Ed. 2013, 52, 10706-10733.PubMed
- Tevyashova AN, Olsufyeva EN, Preobrazhenskaya MN. Design of dual action antibiotics as an approach to search for new promising drugs. Russ Chem Rev. 2015; 84(1): 61CrossRef
- Parkesa AL, Yulea IA. Hybrid antibiotics – clinical progress and novel designs. Expert Opinion on Drug Discovery. 2016; 11(7): 665-680.CrossRef PubMed
- Sampat BN. Academic patents and access to medicines in developing countries. Am J Public Health. 2009; 99(1): 9-17.CrossRef PubMed
- Bern C. Chagas’ disease. N Engl J Med. 2015; 373: 456-466.PubMed
- Apt W. Current and developing therapeutic agents in the treatment of Chagas disease. Drug Des Devel Ther. 2010; 4: 243-253.CrossRef PubMed
- Chatelain E. Chagas disease drug discovery: toward a new era. J Biomol Screen. 2015; 20(1): 22-35.PubMed
- Olmo F, Rotger C, Ramirez-Macias I, et al. Synthesis and biological evaluation of N,N’-squaramides with high in vivo efficacy and low toxicity: towards a low-cost drug against Chagas disease. J Med Chem. 2014; 57: 987-999.PubMed
- Ribeiro CJA, Espadinha M, Machado M, et al. Novel squaramides with in vitro liver stage antiplasmodial activity. Bioorg. Med. Chem. 2016; 24(8): 1786-1792.CrossRef PubMed
- Urbahns K, Härter M, Albers M, et al. Biphenyls as potent vitronectin receptor antagonists. Part 2: biphenylalanine ureas. Bioorg Med Chem Lett. 2003; 13(6): 1071-1074.CrossRef PubMed
- Urbahns K, Härter M, Albers M, et al. Biphenyls as potent vitronectin receptor antagonists. Part 3: Squaric acid amides. Bioorg Med Chem Lett. 2007; 17(22): 6151-6154.CrossRef PubMed
- Raval S, Raval P, Bandyopadhyay D, et al. Design and synthesis of novel 3-hydroxy-cyclobut-3-ene-1,2-dione derivatives as thyroid hormone receptor β (TR-β) selective ligands. Bioorg Med Chem Lett. 2008; 18(14): 3919-3924.CrossRef PubMed
- Jianga CL, Lua CL, Chena YZ, Liub XY. Multiple functions of polypeptides mediated by distinct domains interacting with different receptors. Peptides. 1999; 20(11): 1385-1388.CrossRef
- Neitzel JJ. Enzyme catalysis: the serine proteases. Nature Education. 2010; 3(9): 21.Reference Source
- Zhao G, Perilla JR, Yufenyuy EL, et al. Mature HIV-1 capsid structure by cryo-electron microscopy and all-atom molecular dynamics. Nature. 2013; 497: 643-646.CrossRef PubMed
- Guanga C, Phillips RD, Jianga B, Milanic F. Three key proteases–angiotensin-1-converting enzyme (ACE), ACE2 and renin–within and beyond the renin-angiotensin system. Archives of Cardiovascular Diseases. 2012; 105(6-7): 373-385.Reference Source
- Hunkapiller MW, Forgac MD, Richards JH. Mechanism of action of serine proteases: tetrahedral intermediate and concerted proton transfer. Biochemistry. 1976; 15(25): 5581-5588.CrossRef
- Marciniszyn J, Hartsuck JA, Tang J. Pepstatin inhibition mechanism. Adv Exp Med Biol. 1977; 95: 199-210.CrossRef PubMed
- Nalam MNL, Schiffer CA. New approaches to HIV protease inhibitor drug design II: Testing the substrate envelope hypothesis to avoid drug resistance and discover robust inhibitors. Curr Opin HIV AIDS. 2008; 3(6): 642-646.CrossRef PubMed
- Parai MK, Huggins DJ, Cao H, et al. Design, synthesis, and biological and structural evaluations of novel HIV-1 protease inhibitors to combat drug resistance. J Med Chem. 2012; 55(14): 6328-6341.CrossRef PubMed
- De Clercq E. New developments in anti-HIV chemotherapy. Pure Appl Chem. 2001; 73(1): 55-66.CrossRef
- Bhat TN, Baldwin ET, Liu B, Cheng Y-SE, Erickson JW. Crystal structure of a tethered dimer of HIV-1 proteinase complexed with an inhibitor. Nat Struct Biol. 1994; 1: 552-556.CrossRef PubMed
- Brik A, Wong C-H. HIV-1 protease: mechanism and drug discovery. Org Biomol Chem. 2003; 1: 5-14.CrossRef PubMed
- Fear G, Komarnytsky S, Raskin I. Protease inhibitors and their peptidomimetic derivatives as potential drugs. Pharmacol Ther. 2007; 113: 354-368.CrossRef PubMed
- Kumari G, Singh RK. Highly active antiretroviral therapy for treatment of HIV/AIDS patients: Current status and future prospects and the Indian scenario. HIV AIDS Rev. 2012; 11(1): 5-14.Reference Source
- Lv Z, Chu Y, Wang Y. HIV protease inhibitors: a review of molecular selectivity and toxicity. HIV AIDS (Auckl). 2015; 7: 95-104.PubMed
- Nguyen J-T, Hamada Y, Kiso Y. Design of protein aspartic protease inhibitors to treat various diseases. Arch Pharm Chem Life Sci. 2008; 341(9): 523-535.Reference Source
- Otlewski J, Jelen F, Zakrzewska M, Oleksy A. The many faces of protease-protein inhibitor interaction. EMBO J. 2005; 24(7): 1303-1310.PubMed
- Ghosh AK, Anderson DD. Tetrahydrofuran, tetrahydropyran, triazoles and related heterocyclic derivatives as HIV protease inhibitors. Future Med Chem. 2011; 3(9): 1181-1197.CrossRef PubMed
- De Clercq E. Antiviral drugs: current state of the art. J Clin Virol. 2001, 22(1), 73-89.CrossRef PubMed
- Hodge CN, Aldrich PE, Bacheler LT, et al. Improved cyclic urea inhibitors of the HIV-1 protease: synthesis, potency, resistance profile, human pharmacokinetics and X-ray crystal structure of DMP 450. Chem Biol. 1996, 3(4): 301-314.CrossRef
- Yadav D, Paliwal S, Yadav R, Pal M, Pandey A. Identification of novel HIV 1 – protease inhibitors: application of ligand and structure based pharmacophore mapping and virtual screening. Plos One. 2012; 7(11): e48942.CrossRef PubMed
- Marengo B, De Ciucis C, Verzola D, et al. Mechanisms of BSO (L-buthionine-S,R-sulfoximine)-induced cytotoxic effects in neuroblastoma. Free Radic Biol Med. 2008; 44(3): 474-482.CrossRef PubMed
- Griffith OW. Mechanism of action, metabolism, and toxicity of buthionine sulfoximine and its higher homologs, potent inhibitors of glutathione synthesis. J Biol Chem. 1982; 257(22): 13704-13712.PubMed
- Hanessiana S, Vincia V, Auzzasa L, Marzic M, Gianninic G. Exploring alternative Zn-binding groups in the design of HDAC inhibitors: squaric acid, N-hydroxyurea, and oxazoline analogues of SAHA. Bioorg Med Chem Lett. 2006; 16(18); 4784-4787.CrossRef PubMed
- Volmar C-H, Wahlestedt C. Histone deacetylases (HDACs) and brain function. Neuroepigenetics. 2015; 1: 20-27.CrossRef
- Nishida N, Yano H, Nishida T, Kamura T, Kojiro M. Angiogenesis in cancer. Vas Health Risk Manag. 2006; 2(3): 213-219. PubMed
- Verma RP, Hansch C. Matrix metalloproteinases (MMPs): Chemical-biological functions and (Q)SARs. Bioorg Med Chem. 2007; 15: 2223-2268.CrossRef PubMed
- Diener HC, Kaube H, Limmroth V. Antimigraine drugs. J Neurol. 1999; 246(7): 515-519.CrossRef PubMed
- Saxena PR, Den Boer MO. Pharmacology of antimigraine drugs. J Neurol.1991; 238(Suppl 1): S28-35.CrossRef PubMed
- Sabatoa D, Lionettob L, Martelletti P. The therapeutic potential of novel anti-migraine acute therapies. Expert Opin Invest Drugs. 2015; 24(2): 141-144.CrossRef PubMed
- Kitson SL. 5-hydroxytryptamine (5-HT) receptor ligands. Curr Pharm Des. 2007; 13(25): 2621-2637.CrossRef PubMed
- Humphrey PP. 5-Hydroxytryptamine and the pathophysiology of migraine. J Neurol. 1991;238 (Suppl 1): S38-44.CrossRef PubMed
- Nikolic K, Mavridis L, Djikic T, et al. Drug design for CNS diseases: polypharmacological profiling of compounds using cheminformatic, 3D-QSAR and virtual screening methodologies. Front Neurosci. 2016; 10: 265.CrossRef PubMed
- Kayser V, Aubel B, Hamon M, Bourgoin S. The antimigraine 5-HT1B/1D receptor agonists, sumatriptan, zolmitriptan and dihydroergotamine, attenuate pain-related behaviour in a rat model of trigeminal neuropathic pain. Br J Pharmacol. 2002; 137(8): 1287-1297.PubMed
- Saxena PR, De Vries P, Heiligers JP, et al. BMS-181885, a 5-HT1B/1D receptor ligand, in experimental models predictive of antimigraine activity and coronary side-effect potential. Eur J Pharmacol. 1998; 351(3): 329-339.CrossRef PubMed
- Palló A, Oláh J, Gráczer E. Structural and energetic basis of isopropylmalate dehydrogenase enzyme catalysis. FEBS Journal. 2014; 281(22): 5063-5074.PubMed
- Zhang T, Koshland DE. Modeling substrate binding in thermus thermophilus isopropylmalate dehydrogenase. Protein Sci. 1995; 4(1): 84-92.CrossRef PubMed
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2016; 66(1): 7-30.CrossRef PubMed
- Siddiqui M, Rajkumar SV. The high cost of cancer drugs and what we can do about it. Mayo Clin Proc. 2012; 87(10):935-943.CrossRef PubMed
- Principe P, Braquet P. Advances in ether phospholipids treatment of cancer. Crit Rev Oncol Hematol. 1995; 18: 155-178.CrossRef PubMed
- Farooqui AA, Horrocks LA. Plasmalogens, platelet-activating factor, and other ether glycerolipids. In: Bioactive Lipids. pp. 107-134. (edited by A. Nicolaou and G. Kokotos, The Oily Press, Bridgwater) (2004).Reference Source
- Montrucchio G, Alloatti G, Camussi G. Role of platelet-activating factor in cardiovascular pathophysiology. Physiol Rev. 2000; 80(4): 1669-1699.PubMed
- Wissner A, Kohler CA, Goldstein BM. Analogues of platelet activating factor. 3. Replacement of the phosphate moiety with a sulfonylbismethylene group. J Med Chem. 1985; 28(9): 1365-1367.PubMed
- Elliott TS, Slowey A, Yeb Y, Conway SJ. The use of phosphate bioisosteres in medicinal chemistry and chemical biology. Med Chem Commun. 2012; 3: 735-751.CrossRef
- Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003; 22: 7369-7375.CrossRef PubMed
- Dutton GT. Developmental aspects of drug conjugation, with special reference to glucuronidation. Annu Rev Pharmacol Toxicol. 1978; 18: 17-35.CrossRef PubMed
- Kaivosaari S, Finel M, Koskinen M. N-glucuronidation of drugs and other xenobiotics by human and animal UDP-glucuronosyltransferases. Xenobiotica. 2011; 41(8): 652-69.CrossRef PubMed
- Liston HL, Markowitz JS, DeVane CL. Drug glucuronidation in clinical psychopharmacology. J Clin Psychopharmacol. 2001; 21(5): 500-515.CrossRef PubMed
- Camarasa MJ, Fernandez-Resa P, Garcia-Lopez MT. Uridine 5′-diphosphate glucose analogs. Inhibitors of protein glycosylation that show antiviral activity. J Med Chem. 1985; 28(1): 40-46.CrossRef PubMed
- Vaghefi MM, Bernacki RJ, Dalley NK, Wilson BE, Robins RK. Synthesis of glycopyranosylphosphonate analogs of certain natural nucleoside diphosphate sugars as potential inhibitors of glycosyltransferases. J Med Chem. 1987; 30(8): 1383-1391.CrossRef PubMed
- Kang HT, Hwang ES. 2-Deoxyglucose: An anticancer and antiviral therapeutic, but not anymore a low glucose mimetic. Life Sci. 2006; 78: 1392-1399.CrossRef PubMed
- Davis BG. Recent developments in oligosaccharide synthesis. J Chem Soc Perkin Trans 1. 2000: 2137-2160.CrossRef
- Kamath VP, Diedrich P, Hindsgaul O. Use of diethyl squarate for the coupling of oligosaccharide amines to carrier proteins and characterization of the resulting neoglycoproteins by MALDI-TOF mass spectrometry. Glycoconj J. 1996; 13(2): 315-319.CrossRef PubMed
- Storer RI, Aciroa C, Jones LH. Squaramides: physical properties, synthesis and applications. Chem Soc Rev. 2011; 40: 2330-2346.CrossRef PubMed
- Wurm F, Steinbacha T, Klok H-A. One-pot squaric acid diester mediated aqueous protein conjugation. Chem Commun. 2013; 49: 7815-7817.CrossRef PubMed
- Tietze LF, Schröter C, Gabius S, Brinck U, Goerlach-Graw A, Gabius HJ. Conjugation of paminophenyl glycosides with squaric acid diester to a carrier protein and the use of neoglycoprotein in the histochemical detection of lectins. Bioconjugate Chem. 1991; 2: 148-153.PubMed
- Saksena R, Zhang J, Kováč P. Immunogens from a synthetic hexasaccharide fragment of the O-SP of Vibrio cholerae O:1, serotype Ogawa. Tetrahedron: Asymmetry. 2005; 16(1): 187-197.CrossRef
- Xu P, Alam MM, Kalsy A, et al. Simple, Direct conjugation of bacterial O-SP–core antigens to proteins: development of cholera conjugate vaccines. Bioconjugate Chem. 2011; 22(10): 2179-2185.PubMed
- Alegre-Requena JV, Marqués-Lópeza E, Herrera RP. One-pot synthesis of unsymmetrical squaramides. RSC Adv. 2015; 5: 33450-33462.CrossRef
- Ali TE. Synthetic methods of cyclic α-aminophosphonic acids and their esters. ARKIVOC. 2014; (i): 21-91.Reference Source
- Naydenova E, Troev K, Topashka-Ancheva M, Hägele G, Ivanov I, Kril A. Synthesis, cytotoxicity and clastogenicity of novel α-aminophosphonic acids. Amino Acids. 2007; 33(4): 695-702.CrossRef PubMed
- Franz RG. Comparisons of pKa and log P values of some carboxylic and phosphonic acids: Synthesis and measurement. AAPS PharmSci. 2001; 3(2): 1-13.CrossRef PubMed
- Lavielle G, Hautefaye P, Schaeffer C, Boutin JA, Cudennec CA, Pierre A. New alpha.-amino phosphonic acid derivatives of vinblastine: chemistry and antitumor activity. J Med Chem. 1991; 34(7); 1998-2003.CrossRef PubMed
- Camp NP, Hawkins PCD, Hitchcocka PB, Gani D. Synthesis of stereochemically defined phosphonamidate-containing peptides: Inhibitors for the HIV-1 proteinase. Bioorg Med Chem Lett. 1992; 2(9): 1047-1052.CrossRef
- Hirschmann R, Smith AB, Taylor CM, et al. Peptide syntheses catalyzed by an antibody containing a binding site for variable amino acids. Science. 1994; 265: 234-237.PubMed
- Allen MC, Fuhrer W, Tuck B, Wade RW, Wood JM. Renin inhibitors. Synthesis of transition-state analog inhibitors containing phosphorus acid derivatives at the scissile bond. J Med Chem. 1989; 32(7): 1652-1661.CrossRef PubMed
- Atherton FR, Hassall CH, Lambert RW. Synthesis and structure-activity relationships of antibacterial phosphonopeptides incorporating (1-aminoethyl)phosphonic acid and (aminomethyl)phosphonic acid. J Med Chem. 1986; 29(1): 29-40.CrossRef PubMed
- Schramm VL. Transition states, analogues and drug development. ACS Chem Biol. 2013; 8(1): 71-81.CrossRef PubMed
- Choudhury D, Thompson A, Stojanoff V, et al. X-ray structure of the FimC-FimH chaperone-adhesin complex from uropathogenic escherichia coli. Science. 1999; 285 (5430): 1061-1066.CrossRef PubMed
- Vizcarra IA, Hosseini V, Kollmannsberger P, et al. How type 1 fimbriae help escherichia coli to evade extracellular antibiotics. Sci Rep. 2016; 6: 18109.CrossRef PubMed
- Berry AA, Yang Y, Pakharukova N, et al. Structural insight into host recognition by aggregative adherence Fimbriae of enteroaggregative escherichia coli. PLoS Pathog. 2014; 10(9): e1004404.CrossRef PubMed
- Linhorst TK, Bruegge K, Fuchs A, Sperling O. A bivalent glycopeptide to target two putative carbohydrate binding sites on FimH. Beilstein J Org Chem. 2010; 6: 801-809.PubMed
- Lennox KA, Behlke MA. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011; 18:1111-1120.CrossRef PubMed
- Hunter T. Why nature chose phosphate to modify proteins. Phil Trans R Soc B. 2012; 367: 2513-2516.CrossRef PubMed
- Sato K, Seio K, Sekine M. Synthesis and properties of a new oligonucleotide analogue containing an internucleotide squaryl amide linkage. Nucleic Acids Res. 2001; Suppl(1): 121-122.CrossRef PubMed
- Schmiegelow K, Nielsen SN, Frandsen TL, Nersting, J. Mercaptopurine/methotrexate maintenance therapy of childhood acute lymphoblastic leukemia: clinical facts and fiction. J Pediatr Hematol Oncol. 2014; 36(7): 503-517.PubMed
- Nelson JA, Carpenter JW, Rose LM, Adamson DJ. Mechanisms of action of 6-thioguanine, 6-mercaptopurine, and 8-azaguanine. Cancer Res. 1975; 35(10): 2872-2878.PubMed
- Rudolph J, Stubbe J. Investigation of the mechanism of phosphoribosylamine transfer from glutamine phosphoribosylpyrophosphate amidotransferase to glycinamide ribonucleotide synthetase. Biochemistry. 1995; 34(7): 2241-2250.CrossRef PubMed
- Niewiadomski S, Beebeejaun Z, Denton H, Smith TK, Morris RJ, Wagner GK. Rationally designed squaryldiamides – a novel class of sugar-nucleotide mimics? Org Biomol Chem. 2010; 8: 3488-3499.CrossRef PubMed
- Norbeck DW, Kramer JB, Lartey PA. Synthesis of an isosteric phosphonate analog of cytidine 5′-monophospho-3-deoxy-D-manno-2-octulosonic acid. J Org Chem. 1987; 52(11): 2174-2179.CrossRef PubMed
- Kitson SL, Cuccurullo V, Ciarmiello A, Salvo D, Mansi L. Clinical applications of positron emission tomography (PET) imaging in medicine: oncology, brain diseases and cardiology. Curr Radiopharm. 2009; 2: 224-253.CrossRef
- Saraste A, Knuuti J. Cardiac PET, CT, and MR: what are the advantages of hybrid imaging? Curr Cardiol Rep. 2012; 14(1): 24-31.CrossRef PubMed
- Mak RH, Digumarthy SR, Muzikansky A, et al. Role of 18F-fluorodeoxyglucose positron emission tomography in predicting epidermal growth factor receptor mutations in non-small cell lung cancer. Oncologist. 2011; 16(3): 319-326.CrossRef PubMed
- Romano M, Buratti E. Florbetapir F-18 for brain imaging of β-amyloid plaques. Drugs Today (Barc). 2013; 49(3): 181-93.PubMed
- Sabri O, Seibyl J, Rowe C, Barthel H. Beta-amyloid imaging with florbetaben. Clin Transl Imaging. 2015; 3(1): 13-26.CrossRef PubMed
- Curtis C, Gamez JE, Singh U, et al. Phase 3 trial of flutemetamol labeled with radioactive fluorine 18 imaging and neuritic plaque density. JAMA Neurol. 2015; 72(3):287-94.CrossRef PubMed
- Vorster M, Modiselle M, Ebenhan T, et al. Fluorine-18-fluoroethylcholine PET/CT in the detection of prostate cancer: a South African experience. Hell J Nucl Med. 2015; 18(1): 53-59.PubMed
- Dhingra VK, Mahajan A, Basu S. Emerging clinical applications of PET based molecular imaging in oncology: the promising future potential for evolving personalized cancer care. Indian J Radiol Imaging. 2015; 25(4): 332-341.PubMed
- Portnow LH, Vaillancourt DE, Okun MS. The history of cerebral PET scanning from physiology to cutting-edge technology. Neurology. 2013; 80(10): 952-956.CrossRef PubMed
- Zaidi H, Prasad R. Advances in multimodality molecular imaging. J Med Phys. 2009; 34(3): 122-128.CrossRef PubMed
- Cherry SR. The 2006 Henry N. Wagner Lecture: of mice and men (and positrons)-advances in PET imaging technology. J Nucl Med. 2006; 47(11): 1735-45.PubMed
- Griffeth LK. Use of PET/CT scanning in cancer patients: technical and practical considerations. Proc (Bayl Univ Med Cent). 2005; 18(4): 321-330.PubMed
- Keppler JS. Federal regulations and reimbursement for PET. J Nucl Med Technol. 2001; 29: 173-179.PubMed
- Berger A. How Does It Work? Positron emission tomography. BMJ. 2003; 326(7404): 1449.PubMed
- Lewellen TK. Recent developments in PET detector technology. Phys Med Biol. 2008; 53(17): R287–R317.CrossRef PubMed
- Sodickson L, Bowman W, Stephenson J, Weinstein R. Single-quantum annihilation of positrons. Phys Rev. 1961; 124, 1851.CrossRef
- Tong S, Alessio AM, Kinahan PE. Image reconstruction for PET/CT scanners: past achievements and future challenges. Imaging Med. 2010; 2(5): 529-545. PubMed
- de Vries EG, de Jong S, Gietema JA. Molecular imaging as a tool for drug development and trial design. J Clin Oncol. 2015; 33(24): 2585-2587.CrossRef PubMed
- van Es SC, Venema CM, Glaudemans AW. Translation of new molecular imaging approaches to the clinical setting: bridging the gap to implementation. J Nucl Med. 2016; 57 Suppl 1: 96S-104S.CrossRef PubMed
- Hooker JM. Modular strategies for PET imaging agents. Curr Opin Chem Biol. 2010; 14(1): 105-111.CrossRef PubMed
- Huiban M, Tredwell M, Mizuta S, et al. A broadly applicable [18F]trifluoromethylation of aryl and heteroaryl iodides for PET imaging. Nat Chem. 2013; 5: 941-944.CrossRef PubMed
- Wang J, Sánchez-Roselló M, Aceña JL, et al. Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001-2011). Chem Rev. 2014; 114(4): 2432-2506.PubMed
- Agdeppa ED, Spilker ME. A Review of imaging agent development. AAPS J. 2009; 11(2): 286-299.CrossRef PubMed
- Yamamoto Y, Kurohara T, Shibuya M. CF3-Substituted semisquarate: a pluripotent building block for the divergent synthesis of trifluoromethylated functional molecules. Chem Commun (Camb). 2015; 51(91): 16357-16360.CrossRef PubMed
- Levin VV, Dilman AD, Belyakov PA, Struchkova MI, Tartakovsky VA. Reaction of the Ruppert–Prakash reagent with perfluorosulfonic acids. J Fluorine Chem. 2009; 130(7): 667-670.Reference Source
- Jauw YWS, Menke-van der Houven van Oordt CW, Hoekstra OS, et al. Immuno-positron emission tomography with zirconium-89-labeled monoclonal antibodies in oncology: What can we learn from initial clinical trials? Front Pharmacol. 2016 ;7: 131.CrossRef PubMed
- Zhang Y, Hong H, Cai W. PET tracers based on zirconium-89. Curr radiopharm. 2011; 4(2): 131-139.CrossRef PubMed
- Zeglis BM, Lewis JS. The bioconjugation and radiosynthesis of 89Zr-DFO-labeled antibodies. J Vis Exp. 2015; 96: 52521.PubMed
- Holland JP, Evans MJ, Rice SL, Wongvipat J, Sawyers CL, Lewis JS. Annotating MYC status with 89Zr-transferrin imaging. Nat Med. 2012; 18: 1586–1591.CrossRef PubMed
- Donnelly PS, Rudd SE, Williams SJ. (2016). Novel imaging composition and uses thereof. WO 2016/058056 A1.Reference Source
- Banerjee SR, Pomper MG. Clinical applications of gallium-68. Appl Radiat Isot. 2013; 76: 2-13.CrossRef PubMed
- Gilland DR, Jaszczak RJ, Turkington TG, Greer KL, Coleman RE. Quantitative SPECT imaging with indium-111. IEEE Trans Nucl Sci. 1991; 38(2): 761-766.CrossRef
- Srinivasarao M, Galliford CV, Low PS. Principles in the design of ligand-targeted cancer therapeutics and imaging agents. Nat Rev Drug Discov. 2015; 14: 203-219.CrossRef PubMed
- Zhou Z, Lu Z-R. Gadolinium-based contrast agents for magnetic resonance cancer imaging. Wiley Interdiscip Rev Nanomed Nanobiotechnol. 2013; 5: 1-18.CrossRef PubMed
- Mitsumori LM, Bhargava P, Essig M, Maki JH. Magnetic resonance imaging using gadolinium-based contrast agents. Top Magn Reson Imaging. 2014; 23(1): 51-69.CrossRef PubMed
- Corsi MD, Elst LV, Muller RN, van Bekkum H, Peters JA. Inulin as a carrier for contrast agents in magnetic resonance imaging. Chem Eur J. 2001; 7(1): 64-71.PubMed
- Friedman SH, DeCamp DL, Sijbesma RP, Srdanov G, Wudl F, Kenyon GL. Inhibition of the HIV-1 protease by fullerene derivatives: model building studies and experimental verification. J Am Chem Soc. 1993; 115(15): 6506-6509.CrossRef PubMed
- Gökçinar E, Klapötke TM. Computational study on nitronium squarate – potential oxidizers for solid rocket propulsion? Turk J Chem. 2010; 34: 953-967.Reference Source
- Junaid M, Almuqri EA, Liu J, Zhang H. Analyses of the binding between water soluble C60 derivatives and potential drug targets through a Molecular Docking Approach. PLoS ONE. 2016; 11(2): e0147761.CrossRef PubMed
- Ramström O, Lehn J-M. Drug discovery by dynamic combinatorial libraries. Nat Rev Drug Discov. 2002; 1: 26-36.PubMed
- Lambert JN, Mitchell JP, Roberts KD. The synthesis of cyclic peptides. J Chem Soc Perkin Trans 1. 2001: 471-484.CrossRef
- Tsuruoka H, Shohda K, Wada T. Sekine M. Synthesis and conformational properties of oligonucleotides incorporating 2‘-O-phosphorylated ribonucleotides as structural motifs of pre-tRNA splicing intermediates. J Org Chem. 2000; 65(22): 7479-7494.PubMed
- Pradere U, Garnier-Amblard EC, Coats SJ, Amblard F, Schinazi RF. Synthesis of nucleoside phosphate and phosphonate prodrugs. Chem Rev. 2014; 114(18): 9154-9218.CrossRef