RESEARCH ARTICLE
Igor D. Grachev 1,*, Miroslava Doder 2, †, David J. Brooks 2,3, Rainer Hinz 4
1 Schering-Plough Research Institute, Kenilworth, NJ, USA
2 MRC Clinical Sciences Centre and Division of Neuroscience, Faculty of Medicine, Imperial College, London, UK
3 Hammersmith Imanet Ltd., GE Healthcare, Hammersmith Hospital, London, UK
4 Wolfson Molecular Imaging Centre, University of Manchester, UK
† deceased
Abstract
Background: This PET study was conducted to investigate the receptor occupancy of 11C-SCH442416 in the human brain and to determine plasma concentrations and dose of preladenant which result in inhibition of 11C-SCH442416 binding to adenosine 2A receptors. Preladenant is a novel non-dopaminergic, high-affinity, and highly selective A2A receptor antagonist being investigated for the management of Parkinson’s disease.
Methods: This was an open-label, single-center, and pharmacokinetic-pharmacodynamic study performed in 18 healthy subjects. All subjects received an intravenous injection of the radiotracer 11C-SCH442416. Thirteen subjects received a single dose of preladenant 10, 50 or 200 mg orally at 1, 6 or 12 hrs prior to radiotracer injection.
Results: A blockade of >80% was reached after 50-200 mg doses of preladenant. The Emax model that predicted plasma concentrations corresponding to 50%, 80%, and 90% receptor occupancy was validated. A 5 mg dose, administered BID, was estimated to provide ≥50% receptor occupancy in approximately 75% of the population for the majority of waking hours (12 hours/day).
Conclusions: Single doses of preladenant were well-tolerated. The Cmax and AUC values of preladenant increased in a dose-related manner. In this study we demonstrated the importance of PET imaging for establishing PK-PD relationships and utilizing this tool in confirming proof-of-target and dose guidance for Phase 2/3 clinical trials.
Keywords: 11C-SCH442416; adenosine A2A receptor; PET; spectral analysis
1. Introduction
Parkinson’s disease (PD) is an age-related, progressive neurodegenerative disease characterized by abnormal gait and posture, resting tremor, loss of balance, bradykinesia, and muscular rigidity. Comorbid alterations in mood [1] and cognition [2] are frequently in evidence in PD patients. In the US and Western Europe, approximately 100,000 to 150,000 new cases of PD are diagnosed each year. The disease typically begins to manifest in individuals between 50 and 60 years of age, although onset at younger ages occurs. Current prevalence of PD is estimated at 1.5 million cases in the US and Europe, with the number of incident cases increasing rapidly as the population ages.
The pathology of PD is characterized by a progressive degeneration of the pigmented neurons in the substantia nigra and the presence of intracytoplasmic inclusions known as Lewy bodies. The selective loss of the nigro-striatal dopaminergic pathway and consequent reduction in dopamine levels of the striatum is responsible for the limb bradykinesia and rigidity associated with the disease. The specific etiology of the disease, however, is multifactorial and a number of susceptibility genes and monogenic causes are now known. Manifestation of symptoms is likely to involve a combination of genetic vulnerability, accelerated aging and epigenetic stimuli such as oxidative stress, mitochondrial dysfunction, and excitotoxicity, which ultimately cause specific deterioration of the nigro-striatal dopaminergic pathway.
A potential approach to the treatment of PD is the use of adenosine receptor antagonists. Preladenant is a novel non-dopaminergic, non-methylxanthine, high-affinity (Ki= 1.1 nM), and highly selective (>1000-fold versus A1, A2B, and A3 receptors) A2A receptor antagonist being investigated for the management of movement disorders, including idiopathic Parkinson’s disease. The purine nucleotide adenosine is a ubiquitous modulator of neuronal function in the central and peripheral nervous systems. Adenosine has been shown to exert its biological action through a class of G-protein coupled receptors of which four subtypes have been cloned to date: A1, A2A, A2B, and A3 [3]. Of these, the A2A [4] receptor is highly concentrated in discrete brain nuclei of the basal ganglia (e.g., caudate nucleus, globus pallidus, nucleus accumbens, and olfactory tubercles) [5,6]. Within the striatum A2A receptors are predominantly localized to the GABAergic striatopallidal enkephalin-expressing neurons, where they are co-localized with dopamine D2 receptors, and to intrastriatal GABAergic recurrent collaterals [7,8].
Blockade of A2A receptors on both of these GABAergic neuronal pathways reduces indirect striato-pallidal output neuron activity, thereby compensating for the overactivity of these cells due to dopamine deficiency in PD [9]. The net result is reduced inhibition of neuronal activity in the thalamus (GABAergic) and the thalamo-cortical (glutamatergic) feedback loop [10,11]. These manifests as a suppression of inappropriate motor behaviors characteristic of PD: bradykinesia and akinesia.
Numerous functional studies support the hypothesis that blockade of striatal A2A receptors may provide relief of PD symptoms. A2A antagonists have been shown to activate dopaminergic pathways and to reverse motor impairment in rodent models of PD [12–14]. In nonhuman primates with basal ganglia lesions, A2A receptor antagonists significantly improve motor function without causing dyskinesia [15–17]. Moreover, since the consequence of A2A blockade does not depend on an intact nigro-striatal dopaminergic pathway, it is anticipated that this strategy may provide an opportunity to significantly delay the onset of dopaminergic therapy.
11C SCH442416, the radiotracer being used in this PET study, is a potent, high affinity (Ki=0.048 nM) that is selective for the A2A receptor. It has greater than 20,000 fold selectivity for A2A over A1, A2B, and A3 receptors. It has been previously shown that this radiotracer exhibits robust specific binding and signal-to-noise ratios and is not significantly metabolised in the brain of nonhuman primates [18–20]. It is anticipated that data from this study will confirm that preladenant induces behavioral changes associated with its occupancy of A2A receptors.
The objectives of the present open-label, single-center, pharmacokinetic (PK)/pharmacodynamic (PD), and adaptive study were to investigate the receptor occupancy of 11C SCH442416 in the human brain and to determine the plasma concentration and dose of preladenant which results in inhibition of 11C-SCH442416 binding to A2A receptors.
2. Materials and methods
2.1. Study design and subjects
This was an open-label, single-center, single-dose, PK/PD, and adaptive design PET study performed in 18 healthy male subjects between the ages of 25 and 50 years (mean=32.5±7.7 years), body weight between 62.8 kg and 103.3 kg (mean = 79.52±11.84 kg), and Body Mass Index ranging from 19.2 to 29.0 kg/m2 (mean = 24.66±2.81 kg/m2) were enrolled into and completed this study.
Inclusion and exclusion criteria were chosen to ensure that a well-defined healthy subject population was included in the study. This included detailed medical history and complete physical examination, laboratory test results within normal range, normal vital signs, and normal or clinically acceptable 12-lead electrocardiogram (ECG). Exclusion criteria included any history or presence of clinically significant local or systemic infectious disease within 4 weeks prior to study drug administration, any significant medical disorder which would have required a physician’s care, any history of seizures or autoimmune disorders, any history of mental instability; blood donation within 3 months before administration; history or presence of drug abuse; smoking more than 10 cigarettes or equivalent /day; subjects who have received radiation exposure (including x-rays) within 12 calendar months prior to the study; subjects with significant anatomical abnormalities noted on the MRI of the brain or any condition which would preclude MRI examination (e.g., implanted metal, severe claustrophobia); individuals who had evidence of only one patent arterial supply to the hand (Allen Test).
An initial cohort of five subjects (Baseline Cohort 1) was administered11C‑SCH442416 with the injected activity ranged from 349 to 381 MBq. Data for one subject could not be used because of an acquisition failure during the PET scan; thus data for four subjects were used as baseline [21]. The next cohort of two subjects (Cohort 2) received preladenant 200 mg orally under fasting conditions 12 hours before the administration of 11C-SCH442416. The following cohort of three subjects (Cohort 3) received preladenant 200 mg 1 hour before the administration of 11C-SCH442416. The next three cohorts, Cohorts 4, 5, and 6, with two subjects each, received preladenant 200 mg, 50 mg, and 10 mg, respectively, all at 6 hours before the administration of 11C-SCH442416. Two subjects were included in Cohort 7; both received preladenant 10 mg, one at 1 hour before the administration of 11C-SCH442416 and the other at 12 hours before the administration of 11C-SCH442416. The PET scan for 1 subject given 200 mg treatment in Cohort 3 was not performed because radiotracer specific activity was below specification. Therefore, PET analysis included 16 subjects (4 baseline subjects with radiotracer alone and 12 subjects treated with various doses of preladenant). Because of estimated radiation exposures, subjects were dosed only once with 11C-SCH442416.
The first dose level was 200 mg or the maximum tolerated dose from the single rising dose study and was chosen to set an upper limit on receptor occupancy. Each of the subsequent lower doses (50 and 10 mg) in the progressive decrease was administered at 1, 6 or 12 hours prior to 11C-SCH442416 PET after the receptor occupancy data of the previously administered higher dose were evaluated. The decision to progress to lower dose levels at different scanning times was determined by review of the PET images and time-activity curves of 11C-SCH442416 activity in the putamen, caudate, thalamus and cerebellum.
Blood PK samples for preladenant plasma concentration assessment were collected pre dose (0 hour), 0.25, 0.5, 0.75, 1, 1.25, 1.5, 2, 3, 4, 5, 6, 8, 10, 12, 18, and 24 hours after dosing of preladenant. Additionally, samples were drawn immediately before the PET scan and at the end of the 90-minute PET study. This information was used to characterize the effect of preladenant on the receptor occupancy profile at 1, 6, or 12 hours post dose. Plasma samples were analyzed using a validated liquid chromatographic method with tandem mass spectrometric detection.
This study was conducted in accordance with the Declaration of Helsinki Principles, ICH guidelines for Good Clinical Practice and after approval was obtained from the Research Ethics Committees at Central Middlesex and Hammersmith Hospitals, London; and the U.K. Administration of Radioactive Substances Advisory Committee. All subjects signed and dated an IEC-approved consent form before being enrolled into the study. All subjects were covered by Health Insurance System and/or in compliance with the recommendations of National Law in force relating to biomedical research.
2.2. PET/MRI scanning protocol
All PET scans were performed on the high-sensitivity Siemens/CTI scanner ECAT EXACT3D with an axial field of view of 23.4 cm and 95 reconstructed transaxial image planes [22]. A 5-minute transmission scan using a 137Cs point source was carried out prior to each study for subsequent attenuation and scatter correction. The 95-minute three-dimensional dynamic emission scan was acquired in list mode. In the post acquisition frame rebinning, 28 time frames of increasing length were generated (30-second background frame prior to the injection, one 15-second frame, one 5-second frame, four 10-second frames, four 60-second frames, and seventeen 300-second frames). The PET images were reconstructed using filtered back projection based on the following set of parameters: scatter correction was model based; attenuation correction was measured and segmented; reconstruction machine was SUN CPU with a zoom 2.5; spatial resolution of reconstructed images was 5.1 mm x 5.1 mm x 5.9 mm; and reconstructed voxel size was 2.10 mm x 2.10 mm x 2.43 mm.
The radiotracer 11C-SCH442416 synthesis was described previously [19] and was injected into an antecubital vein as a smooth bolus over 30 seconds. The mean injected radioactivity dose of 11C-SCH442416 was 362±9 MBq, radiochemical purity of the injected radiotracer at time dispense was > 99%, injected mass of SCH442416 was 2.54±1.1 µg, and specific activity at time of injection was 66.6±31.0 MBq/nmol. The estimated volume for injected radiotracer was 1.91±0.78 mL, and pH of the injected solution was 5.62±0.42.
After i.v. injection of 11C-SCH442416, the radioactivity in blood was measured using a continuous on-line radioactivity detector system [23]. Discrete arterial blood samples were withdrawn before the PET scan (baseline sample) and throughout the PET scans via a cannula inserted into the opposite arm to which the radiotracer was administered. The withdrawal times were to be 5, 10, 15, 20, 30, 40, 50, 60, 75, and 95 minutes post 11C-SCH442416 injection for the first three scans. Beginning with the fourth scan, this sampling schedule was subsequently revised to more frequent sampling at the beginning due to the fast kinetics of 11C-SCH442416.
For each subject, brain MRIs were acquired with T1 weighted RF spoiled gradient echo volume scans on a 1 Tesla Philips Medical Systems HPQ+ Scanner. The echo time TE was 6 ms, the repetition time TR was 21 ms, the flip angle was 35°, yielding an image resolution of 1.6 mm x 1.6 mm x 1.0 mm (AP, LR, and HF, respectively).
A set of 49 volumes of interest (VOI), which included the cerebellum, was defined with the help of a probabilistic brain atlas template [24]. The standard MRI on which the VOI template had been defined was normalized to the individual MRI using the SPM99 software package (Statistical Parametric Mapping, Wellcome Department of Cognitive Neurology, London, United Kingdom). The calculated image transformation parameters were then applied to transform the VOI template, resulting in a map of VOIs of the subject’s individual MRI space. The individual MRI was then coregistered to the PET image summed from 1 to 30 minutes after tracer injection using an automated multiresolution optimization procedure [25]. On the individual MRI, the following six ROIs were defined manually: left and right putamen, left and right caudate, and left and right thalamus. The cerebellum was defined with the help of a probabilistic brain atlas template [24]. For the manual drawing of the ROIs as well as for the generation of the TACs, the medical imaging software package ANALYZE [26,27] was used.
2.3. Primary endpoint and statistical methods
2.3.1. Quantification of radiotracer binding and receptor occupancy
Spectral analysis was used to estimate the regional binding potentials and receptor occupancies. The use of spectral analysis for the quantification of dynamic PET studies was introduced by Cunningham and Jones [28]. In contrast to compartmental modeling, spectral analysis does not define a certain structure of compartments. Spectral analysis is used in PET for the quantification of cerebral glucose metabolism in 18F-FDG studies, for the quantification of cerebral blood flow in scans with 15O-water and for the quantification of neuroreceptor binding, e.g. in studies with the opiate receptor radiotracer 11C-diprenorphine [28]. For a general discussion of the applicability of spectral analysis see the work of Schmidt [29]. For the analysis of the dynamic PET scans with 11C-SCH442416, spectral analysis was used because of its ability to separate three components of the tissue response function: irreversible components = slow nondisplaceable uptake, reversible components = fast nonspecific and specific (displaceable) binding, and fast components = plasma volume. Only the specific binding in blocked scans (reversible components) turned out to be displaceable by the blocking drug. The irreversible uptake did not seem to be affected by the blocker. BP estimates were calculated as VDtarget/VDreference – 1, VD target being the volume of distribution of the reversible components in the target region and VD reference the volume of distribution of the reversible nonspecific binding in the cerebellum.
The mean OCC of a region for each subject was calculated as:
OCC = ( 1 – BPBlocked / BPBaseline ) x 100%
with BP blocked as the binding potential of the subject after administration of blocking compound preladenant, and BP baseline as the mean baseline binding potential in that region.
2.3.2. Pharmacokinetic analysis
Individual plasma concentration-time data were analyzed using model-independent methods [30]. The maximum concentration (Cmax, ng/mL), time of maximum concentration (Tmax, hr), and the time of final quantifiable sample (tf, hr) were the observed values. The area under the plasma concentration-time curve from time 0 to tf (AUC [tf], ng∙hr/mL) was calculated using the linear trapezoidal method. The terminal phase rate constant (K, hr-1) was calculated as the negative of the slope of the terminal log-linear portion of the plasma concentration-time curve using linear regression. The area under the plasma concentration-time curve from Time 0 to infinity (AUC [I], ng∙hr/mL) was calculated as follows:
AUC (I) = AUC (tf) + Cest / K
where Cest is the estimated plasma concentration at tf.
The apparent terminal-phase half-life (t½, hr) was calculated as follows:
t1/2 = In(2) / K
These data are listed in Table 2 and Table 3. PK results were summarized using geometric mean and coefficient of variation for each dose level. Preladenant plasma concentration values were used for PK-PD analyses.
2.3.3. Pharmacokinetic-pharmacodynamic (PK-PD) analysis
Receptor occupancy data from the putamen region of the brain were selected for PK-PD model development due to the high binding potential for A2A receptors and less variation between the unilateral (left and right regional) occupancy values of preladenant in this region of interest (ROI), which is involved in pathophysiology of Parkinson’s disease. Relationship between plasma preladenant concentrations obtained at the start of PET image acquisition and the receptor occupancy in the specific ROI (i.e., putamen) was graphically explored. An Emax model [31] described below was selected to fit the PK-PD data.
E = Emax . C / EC50 + C
where,
E= Receptor occupancy (%)
C= Plasma concentration (ng/mL) corresponding to the receptor occupancy E
Emax= An asymptote representing the maximum receptor occupancy (%)
EC50= Plasma concentration (ng/mL) corresponding to 50% of Emax
Two additional parameters were derived to characterize various levels of receptor occupancy of preladenant:
EC80= Plasma concentration (ng/mL) corresponding to 80% of Emax. The EC80 was calculated directly from eq.1 by substituting E=80% of Emax, and then solving for C.
ECopt= Plasma concentration (ng/mL) corresponding to optimal receptor occupancy. ECopt was defined as the lowest observed plasma concentration that achieved the model estimated Emax (i.e., ≥ 85% occupancy).
In order to obtain population estimates of the duration of receptor occupancy, the available plasma concentration data from five completed Phase 1 studies (n=130) were pooled. Descriptive statistics, such as the10th and 25th percentile, mean, and maximum plasma concentration values were calculated at each time point. Population estimates of receptor occupancy durations were estimated as explained by a specific case below. Following a single oral dose of 25 mg of preladenant, the time duration over which the 10th percentile plasma concentration remained greater than or comparable to the EC50 value, was considered as the population estimate of time duration for 50% receptor occupancy of preladenant in 90% of population. In this manner, prelandenant receptor occupancy durations were determined following various doses and QD or BID regimens of preladenant in ~75% and ~90% of population.
3. Results and discussion
3.1. Summed PET images and time activity curves
A total of 18 healthy subjects completed the study: 13 received oral preladenant before 11C-SCH 442416 PET (Cohorts 2-6) while 5 had 11C-SCH442416 PET alone (Cohort 1). The tracer injected activity ranged from 349 to 381 MBq. Data for one baseline subject were not usable because of an acquisition failure during the PET scan. The PET scan for one subject from the preladenant treatment group had to be canceled, because the radiotracer delivered was below specifications, and thus only 12 subjects were included in the blockade results (Table 1).
Note: Subject No. 3 had equipment error during PET data acquisition, data can not be recovered. Subject No.12 had enlarged ventricles (right > left), which can explain the unusually high occupancy value for the right caudate. Shown average and standard deviation (SD) values.
Table 1. Individual PET Binding Potentials at Baseline, and Binding Potentials and Receptor Occupancies Following Oral Administration of Single 10, 50, or 200 mg Doses of Preladenant at 1, 6 and 12 hours Post Dose.
After intravenous (i.v.) bolus injection of 11C-SCH 442416, the radioactivity appeared rapidly in all studied brain regions, peaking around 3 minutes after injection. A relatively fast reversible component of uptake was apparent in parallel with a slower and irreversible component over the time course of scanning. In target regions, only the reversible uptake component was reduced by pre-administrationt of the selective A2A receptor antagonist preladenant. The cerebellum time activity curve (TAC) was unaffected by preladenant and was used as a reference region for the estimation of nonspecific tracer binding.
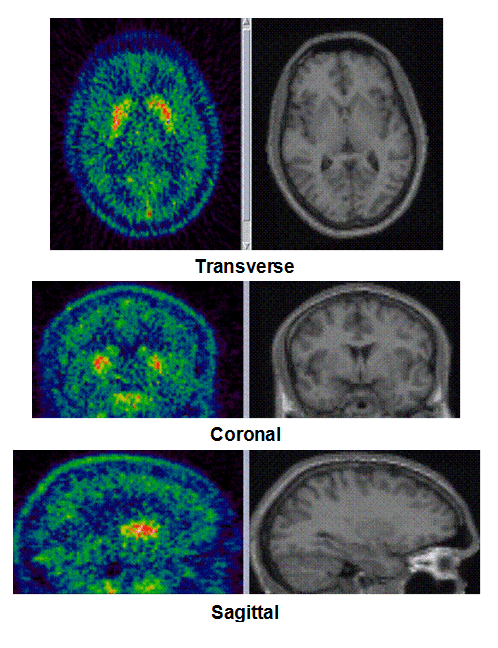
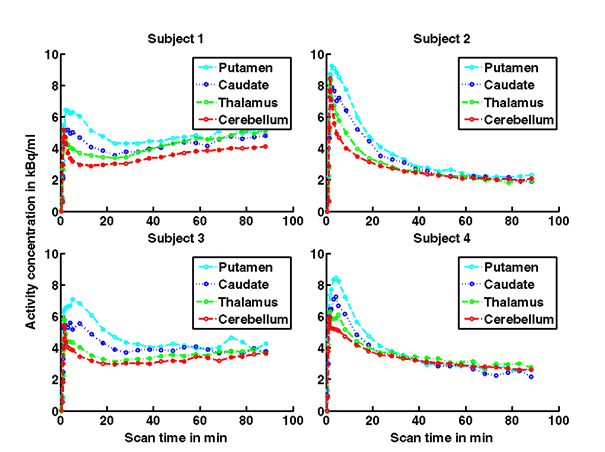
Figure 1a. On the left hand side the added PET images from approximately 1 to 30 minutes after intravenous bolus injection are shown. The color map of the PET images is of type rainbow, ranging from black = 0 kBq /cm3 to red (white) = 5.3 kBq /cm3. On the right hand side the individual MR images are shown. They were co-registered and resliced to the PET image space and are coded in a linear grey scale. The images are displayed in transverse (axial), coronal and sagittal sections. Baseline PET/MRI scans and time activity curves (TACs) for putamen, caudate, thalamus and cerebellum are shown for 4 subjects. PET/MR images and TACs showed the highest activity concentrations in the A2A receptor-rich striatum region.
On the left Figure 1a shows baseline PET images from approximately 1 to 30 minutes after i.v. bolus injection of 11C-SCH442416, and co-registered and resliced individual MR images in transverse (axial), coronal and sagittal views in 4 subjects (baseline cohort); on the right it shows corresponding TACs for putamen, caudate, thalamus and cerebellum. Radiotracer uptake was highest in the A2A receptor-rich striatum (putamen and caudate) region and lowest in the cerebellum and thalamus (Fig. 1a). After administration of the lowest tested dose 10 mg of preladenant (Fig. 1b) the difference in 11C-SCH442416 uptake between striatum and cerebellar regions was reduced (partial blockade shown for 10 mg at 12 hours post dose). After administration of the mid and highest doses (50 and 200 mg) of preladenant regional TACs for striatum regions overlapped with the cerebellum during the entire duration of the PET scan demonstrating a full blockade of A2A receptors (Fig 1c full blockade shown for 50 mg at 6 hours post dose). A rainbow color map scale has been applied to all PET add images ranging from black (0 kBq /cm3) to red/white (5.3 kBq /cm3).
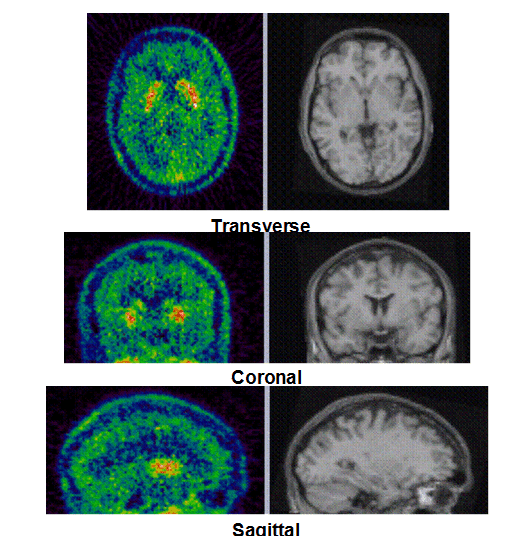
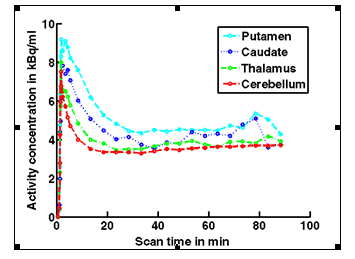
Figure 1b. An example of PET/MRI scans and TACs in subject who received the lowest dose 10 mg of preladenant and had a partially blocked PET scan at 12 hours post dose.
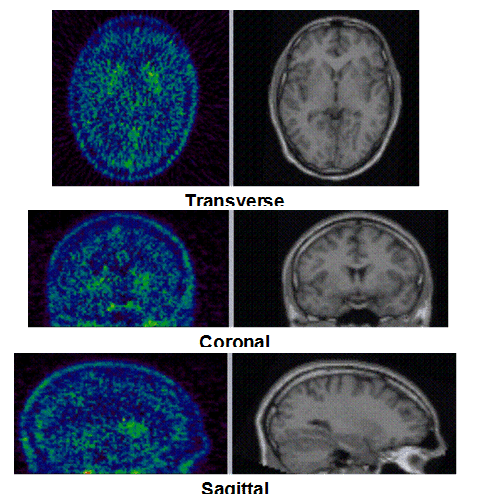
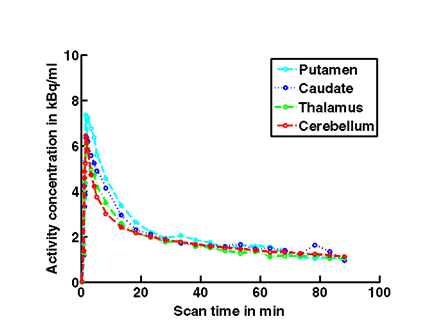
Figure 1c. An example of PET/MRI scans and TACs in subject who received the mid dose 50 mg of preladenant and had a fully blocked PET scan at 6 hours post-dose.
3.2. Binding potential values and receptor occupancy results
Table 1 summarizes the estimated binding potential (BP) values and receptor occupancies (OCC) in the putamen, caudate, thalamus and the cerebellum under baseline and blocked conditions for all subjects. The average BP estimates of the baseline cohort were 2.47±0.84 for putamen (average ± standard deviation [SD]), 1.59±0.70 for caudate, and 0.53±0.48 for thalamus.
The average BP and OCC values in all studied brain regions for each individual subject and dosing-time cohort in the blocking studies were calculated and presented in the Table 1. Administration of 200 mg preladenant led to 88-105% A2A receptor occupancy in the striatum by 11C-SCH442416 and on average 61% occupancy in the thalamus 1 hour after dosing (Table 1). By 6 hours after administration of 200 mg, the A2A occupancy remained at a level of 82-93% in the striatum but fell to 43% in the thalamus. The occupancy 12 hours after a 200 mg dose was reduced to 56-58% in the striatum but remained 43% in the thalamus. One cohort was dosed with 50 mg preladenant: the measured occupancies 6 hours post dose were similar (84-91%) to the 200 mg dosed cohort. Two further cohorts were dosed with 10 mg preladenant: the occupancies in the striatum were measured at 75-78% at 1 hour, 47-53% at 6 hours, and 15-19% at 12 hours post dose (Table 1).
3.3. Pharmacokinetic results
Because of the small number of subjects (n= 4, 2, and 7 for the 10, 50, and 200 mg dose-groups, respectively), only descriptive statistics of the pharmacokinetic parameters were reported. For the parent drug, the mean Cmax values following 10, 50, and 200 mg doses were 111, 434, and 890 ng/mL, respectively, and the mean AUC (tf) values were 275, 1401, and 2597 ng×hr/mL, respectively (Table 2). The Cmax vs dose and AUC (tf) vs dose relationships for the preladenant showed that the increase in Cmax and AUC was less than dose-proportional at the 200 mg dose. The inter-subject variability in plasma preladenant concentrations was high, as reflected in the large coefficient of variation (CV) values reported across the time points. Consequently, the CV values for Cmax and AUC (tf) ranged from 43% to 55% and 46% to 65%, respectively.
Note: NR = Not reported.
Table 2. Mean Pharmacokinetic Parameters of Preladenant Following Oral Administration of Single 10, 50, or 200 mg Doses.
3.4. Pharmacokinetic/pharmacodynamic results
Plasma preladenant concentrations obtained at the start of the 90-minute PET scan and the corresponding receptor occupancy values are presented in Table 1 along with the Emax model-predicted data, which are shown in the Figure 2. A good fit of the Emax predicted to the observed data was demonstrated by an r-value of 0.84 and the distribution of the residuals (observed minus predicted values) around the line of identity. No systematic deviations were observed in the residuals.
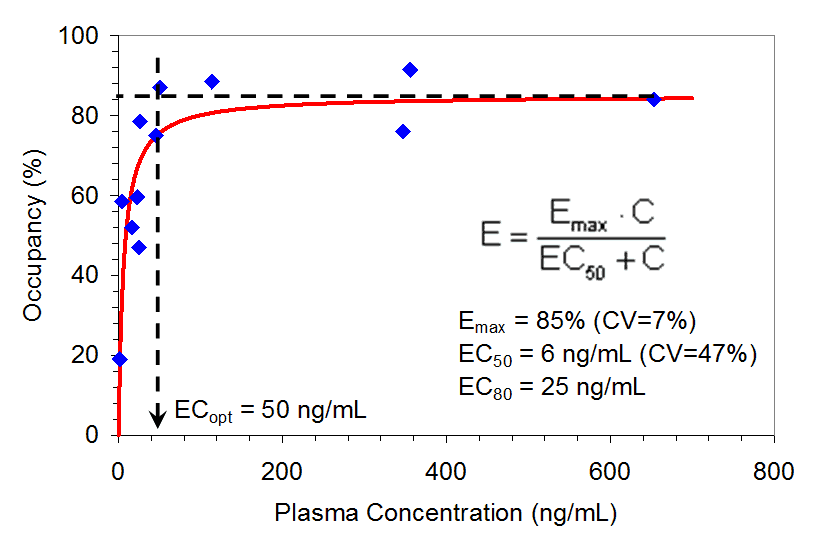
Figure 2. Relationship between A2a Receptor Occupancy in the Putamen Region of the Brain and Plasma Preladenant Concentrations (n= 12). PET scan for one subject was canceled, tracer was below specifications, and thus only 12 subjects were included.
Receptor occupancy increased rapidly as plasma concentrations increased, and attained a maximum occupancy at around 50 ng/mL. This maximum was maintained over a wide range of plasma concentrations (> 600 ng/mL). The model-predicted parameter estimates were as follows:
Emax = 85% (CV=7%), EC50 = 6 ng/mL (CV=47%), EC80 = 25 ng/mL and ECoptimal(ECopt) = 50 ng/mL.
The CV for EC50 was large (47%) owing to the steep slope of the ascending portion of the receptor occupancy-plasma concentration curve and the sparse data in this portion of the curve. ECopt was estimated in model-independent manner. However, the plasma concentration representing ECopt also represented 90% of the model predicted Emax, thus providing a good validation of model-based predictions.
The PK-PD relationship was used to assist the selection of clinical doses for the Phase 2 studies (Figure 2). The time durations following the 50 mg dose for ³50%, ³80%, and ³90% receptor occupancy were estimated to be 10, 5, and 4 hours, respectively, in approximately 90% of the population, and 24, 6, and 5 hours, respectively, in approximately 75% of the population (Figure 3).
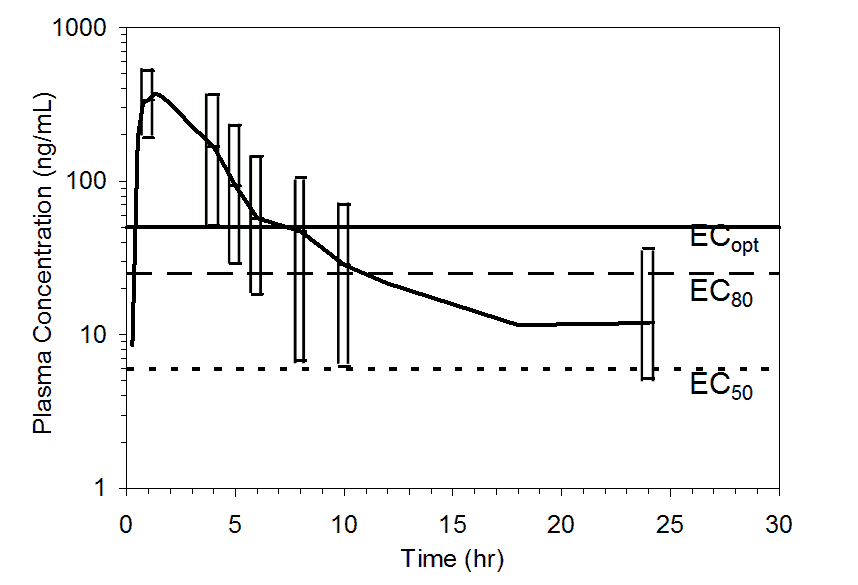
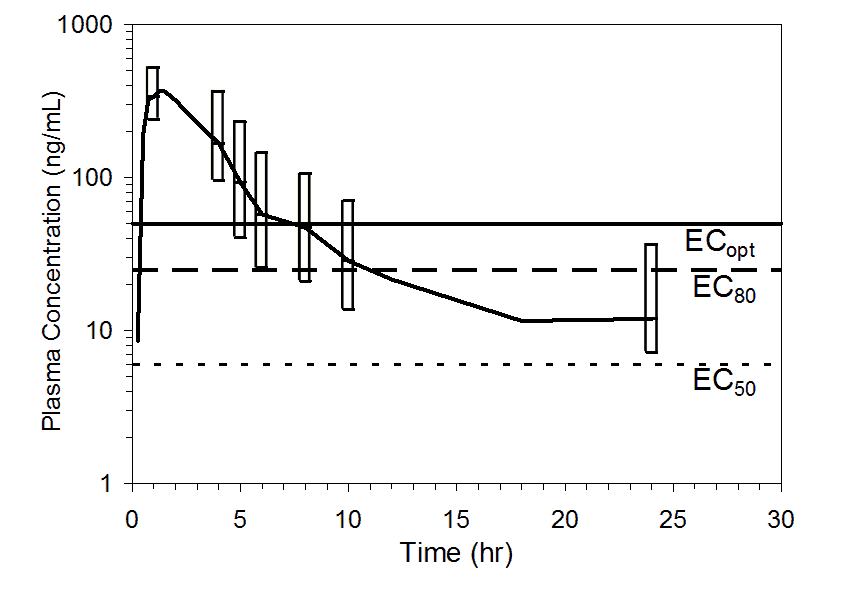
Figure 3. Mean pooled plasma preladenant concentrations following administration of a single 50 mg oral dose of preladenant (n= 17). Top figure shows upper 90% (10th percentile to maximum), and the bottom figure shows upper 75% (25th percentile to maximum) ranges at 1, 4, 5, 6, 8, 10, and 24 hours. Plasma concentration data were pooled from five completed Phase 1 PK studies. Threshold plasma concentrations representing EC50, EC80, and ECopt are overlaid to illustrate how durations of receptor occupancy were determined.
The time durations were thus estimated for single doses of preladenant ranging from 5 to 100 mg (Table 3). Following a single 5 mg dose, receptor occupancy of ³80% for approximately 2 hours is expected to be attained in approximately 75% of the population and receptor occupancy of ³50% for approximately 6 hours in approximately 75% of the population. In contrast, a single 100 mg dose is expected to provide ³90% receptor occupancy for approximately 4 hours in approximately 90% of the population, and for about 6 hours in approximately 75% of the population as illustrated in Table 4 and Figure 4.
Table 3. Individual Subject Plasma Preladenant Plasma Concentrations and Receptor Occupancy Values for the Putamen Region of Interest.
Note:
a: For each dose, plasma concentration data were pooled from the 5 completed Phase 1 studies of preladenant.
b: Time duration over which plasma concentrations remain ³50 ng/mL.
c: Time duration over which plasma concentrations remain ³25 ng/mL.
d: Time duration over which plasma concentrations remain ³6 ng/mL.
e: Following 100 mg dose, 87% of population is estimated to exhibit ³50% receptor occupancy up to 24 hours postdose.
Table 4. Predicted Time Durations of A2A Receptor Occupancy for the Putamen Following Single Doses of Preladenant Based on Population PK Data Pooled from Five Completed Phase 1 studies.
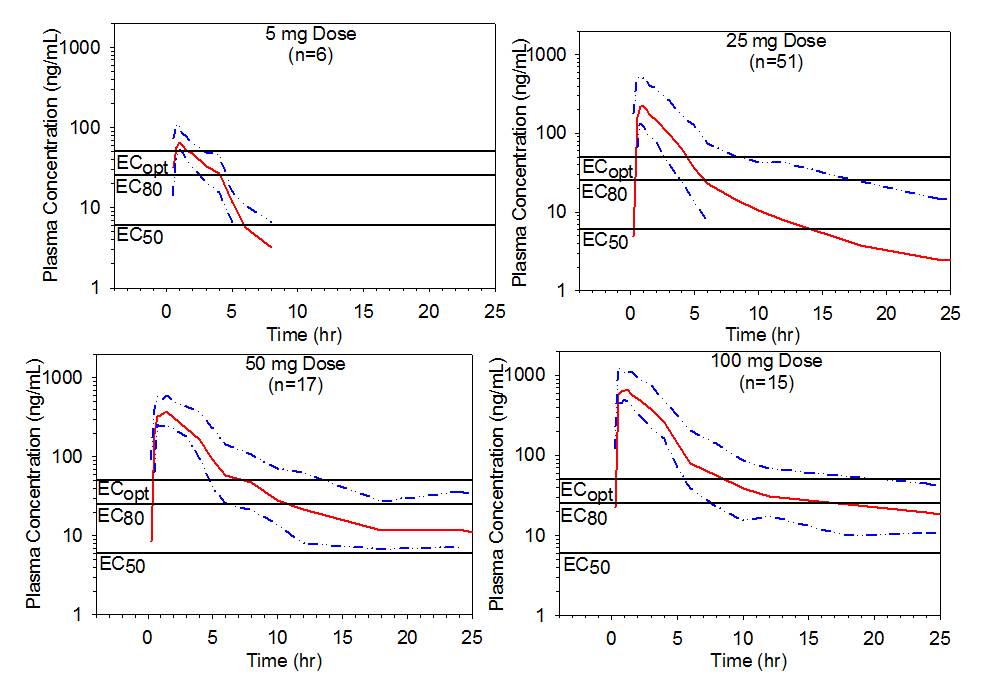
Figure 4. Mean pooled plasma preladenant concentration-time profiles with upper 75% range following single oral doses of 5, 25, 50, or 100 mg of preladenant. Threshold plasma concentrations representing EC50, EC80, and ECopt are overlaid to illustrate how durations of receptor occupancy were determined.
This procedure was further extended to include the multiple dose scenarios, using simulations by superposition of the pooled plasma profiles following different doses and dosing regimens of preladenant (Table 5, Figure 4 and Figure 5). The predictions indicated that the duration of receptor occupancy increased with the dose and dosing frequency. Although a 5 mg dose reached ³80% receptor occupancy following BID administration, the duration of occupancy was short and was predicted to increase to a total of 8 hours per day with QID administration in approximately 75% of the population. Results of this study also suggest that longer duration of ³50% receptor occupancy following 5 mg BID administration can be achieved in approximately 75% of the population for 12 hours/day, which represents 67% (12/18 hours) of waking time. Conversely, a 50 mg BID administration was predicted to provide ³80% receptor occupancy for about 18 hours per day in approximately 75% of the population. Assuming that 80% or higher receptor occupancy would be clinically desirable, a 50 mg dose administered twice daily, 8 hours apart, appeared to meet this criterion during the waking hours in the majority (³75%) of the population. Moreover, a 50 mg QID administration was predicted to provide ³90% receptor occupancy over a 24-hour period in approximately 75% of the population.
Note: a: Pooled from five completed Phase 1 PK studies of preladenant.
Table 5. Predicted durations (hr/day) for A2A receptor occupancy for the putamen following various regimens of 5 mg, 25 mg, and 50 mg doses of preladenant based on population PK data pooled from five completed Phase 1 studies.
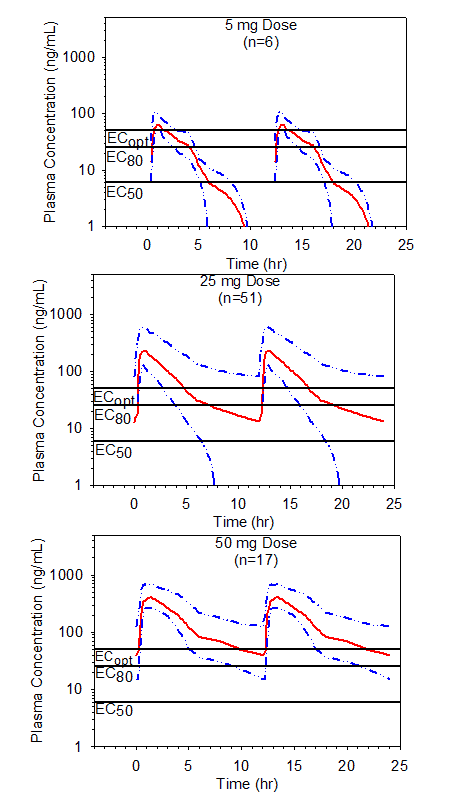
Figure 5. Simulated mean steady state plasma preladenant concentrations following BID single dose administration of 5, 25 or 50 mg doses of preladenant. Data shown with the upper 75% (25th percentile to maximum) ranges. Threshold plasma concentrations representing EC50, EC80, and ECopt are overlaid to illustrate how durations of receptor occupancy were determined.
3.5. Safety
There were no serious adverse events or deaths, and no subject discontinued this study because of an adverse event (AE). Six out of 18 (33%) subjects reported at least one treatment-emergent AE: 5 out of 13 (38%) subjects treated with preladenant and 1 out of 5 (20%) subjects who received only 11C-SCH 442416 radiotracer. AEs reported by subjects treated with preladenant included dizziness (reported by 1 subject treated with preladenant 50 mg), headache (reported by 1 subject treated with preladenant 10 mg, and 1 subject treated with 200 mg dose), syncope (reported by 1 subject treated with preladenant 50 mg), and vasovagal attack and anxiety (each reported by 1 subject treated with preladenant 200 mg). Mild dizziness, considered by the investigator to be unrelated to treatment with study drug, was the only AE reported in subjects exposed to 11C-SCH 442416. The majority of AEs were mild in severity. There were no clinically significant changes in vital signs or ECG parameters including QTc and PR intervals. There were no clinically significant abnormalities noted in hematology, blood chemistry, or urinalysis.
3.6. Discussion
PET is commonly viewed as a well-established, promising and highly sensitive technology in CNS drug development, which provides important information related to target engagement (proof-of-target), mechanism of action (proof-of-mechanism), biological effect (proof-of-biology), pharmacological effect (proof-of-pharmacology), required level of receptor block/occupancy (proof-of-occupancy), and aids the Pharmaceutical industry for go/no-go decision-making and rationalizing dose-range selection for Phase 2b and 3 clinical trials. Quantitative in vivo imaging of A2A receptors in the human brain using 11C-SCH442416 PET has been recently described [21].
The current study was designed to investigate the A2A dose-receptor occupancy profile of preladenant in the human brain and to determine plasma concentrations and dose of preladenant that result in inhibition of 11C-SCH 442416 binding at 1, 6 or 12 hours after single oral administration of 10, 50 and 200 mg. The BPs of 11C-SCH 442416 in the putamen, caudate, and thalamus at baseline, and after blockade with the selective A2A receptor antagonist preladenant were determined. The BP is a measurement of the specific radiotracer 11C-SCH 442416 volume of distribution and reflects the ratio of the density of available receptors and their affinity for the radiotracer. Before administration of preladenant there should be a high capacity of receptors for binding, so the BP will be high. After administration of preladenant the study drug will occupy many of the available receptors, so there will be a decreased capacity of receptors for radiotracer binding and the BP will be lower. BP is calculated as the ratio of bound to free radiotracer at equilibrium. The calculation of the BP requires an estimate of non-specific region binding, typically obtained from a separate reference region with no specific binding – in this case the cerebellum – assumed to be representative.
The blocking of 11C-SCH442416 binding following administration of preladenant was investigated in three target ROIs namely putamen, caudate and thalamus with primary emphasis on the putamen in exploratory model-predicted analyses. The cerebellum was used to estimate non-specific binding since it has been reported that there is little specific A2A binding and the lowest radiotracer uptake is found in this region possibly reflecting a low density of A2A receptors [32,33]. The BP values were consistently higher in the striatum when compared to the thalamus and cerebellum with a small intersubject variability consistent with the findings of Mishina et al. [32] and Naganawa et al. [33] who reported the highest radioactivity concentrations in the striatum in the human brain using the A2A marker 11C-TMSX PET. The observed BP values were also consistent with those reported in vivo in animal studies [20] and postmortem human brain distribution studies of A2A receptors using whole hemisphere autoradiography and 3H-CGS 21680 [34].
Negative BP values were observed in the caudate region for the blocked PET images for those subjects who received the highest dose (50 and 200 mg) after shortest time (1 and 6 hrs) post treatment. From the time activity curves for these subjects it is clear that the fully blocked caudate region shows a lower uptake over the course of the PET scan than the cerebellum for 50-200 mg at 1-6 hours post dose. The presence of negative BP values in fully blocked scans is most likely a consequence of the underestimation of the activity in the head of caudate due to its proximity to the ventricle and the limited spatial resolution of PET (partial volume effect). It may also reflect the fact that the cerebellum is not a true reference region for the non-specific tracer binding, overestimating its contribution. Their presence could also reflect the fact that baseline scans were obtained from different subjects to those with blocked scans due to local limits for allowable radiation exposure in the first-in-man evaluation of the novel radiotracer. Finally, while spectral analysis separates three frequency components of the tissue response function model in unblocked subjects, the tracer kinetics are changed in the blocked situation.
Negative BPs are not uncommon and have been reported in other full blocking studies with other tracers [35,36]. A blockade of >80% was reached with 50-200 mg dose of preladenant either at 1 hour and continued up to 6 hours post dose. As the blocking dose of preladenant is decreased the BP values increased as more of the receptors became available to bind the radiotracer.11C-SCH442416 uptake was highest in striatal regions and their blocked BPs increased as the dose of preladenant was decreased. We found that the A2A receptor occupancy decreased from a full blockade at 1-6 hours to 56-58% at 12 hours after 200 mg, and demonstrated dose- and time-dependent decreases as the dose was decreased to 10 mg preladenant and time increased from 1 to 6 or 12 hours respectively.
The mean exposure to preladenant increased as the dose increased for all dose groups. Variability in exposure was relatively high. Preladenant was rapidly absorbed following oral administration with the mean Tmax occurring between 1.0 to 1.38 hours post dose. The terminal elimination half-life was between 2.14 and 4.56 hours. The PK data presented suggest that the human PK is predictable in the dose range studied and when combined with PET receptor occupancy data can be used as a valuable tool to determine the plasma concentration/receptor occupancy relationship.
In this study we established the relationship between A2A receptor occupancy (pharmacodynamic [PD] parameter) and plasma concentration (PK parameter) of preladenant, and the dose of preladenant. This PK-PD relationship was described by an Emax mixed effect model, which allowed us to estimate predicted preladenant plasma concentrations corresponding to different levels of theoretical receptor occupancy at various times post dose. The PK-PD model makes the simplifying assumption that any metabolites of preladenant contribute minimally to A2A receptor occupancy (there is no evidence that metabolites cross the blood brain barrier in humans and show central activity in pharmacological models) and there is a rapid equilibrium between plasma concentration and receptor binding (i.e., on and off-rates) in the brain. The theoretical predictions from this model provide an attractive decision-making approach for providing information on the appropriate dose range for Phase 2 and 3 studies. When combined with available safety data, an overall profile regarding PK, safety and activity (based upon receptor occupancy) can be made providing information on the potential therapeutic dose range.
To date, there have been no receptor occupancy PET results available for preladenant using the A2A specific radiotracer 11C-SCH442416. Blocking studies with experimental adenosine A2A antagonists KF15372 and ZM 241385 failed to demonstrate an inhibition of A2A radiotracer 11C-KW-6002 in rats [37]. A2A occupancy by ASP5854 (0.001–0.1 mg/kg) was examined in the striatum using11C-SCH442416 PET in conscious rhesus monkeys [20]. After administration of ASP5854 the maximum receptor occupancy was estimated to be 85-90% [20]. One study using 11C-SCH442416 PET and a non-xanthine, selective A2A receptor antagonist in development, vipadenant (previously known as BIIB014) has been reported [38]. In this study the estimated receptor occupancy of vipadenant in the brain regions of interest varied from 74% to 94% at the lowest daily dose (2.5 mg) and reached saturation in all regions at 100 mg. A similar range for the maximum receptor occupancy was demonstrated in this first human dose-occupancy study.
Based on PK-PD modeling we used prior knowledge from five completed Phase 1 PK studies with estimated plasma concentration vs. time relationships to predict duration of A2A receptor occupancy following single dose administration theoretical scenarios which can enable us to characterize a time-on-target and duration of receptor occupancy. Projections for predicted receptor occupancy over the 5 to 100 mg dose range over 24 hours were generated to better understand single dose predicted occupancy-time profiles for all studied dose levels of preladenant. These projections indicate that following a single 5 mg dose, receptor occupancy of ³80% for approximately 2 hours and of ³50% for approximately 6 hours are expected to be attained in approximately 75% of the population. In contrast, a single 100 mg dose is expected to provide ³90% receptor occupancy for approximately 4 hours in approximately 90% of the population, and for about 6 hours in approximately 75% of the population.
This approach was further extended to include the multiple dose scenarios, using simulations by superposition of the pooled plasma profiles following different doses and dosing regimens of preladenant. The predictions indicated that the duration of receptor occupancy increased with the dose and dosing frequency. Although a 5 mg dose reached ³80% receptor occupancy following BID administration, the duration of occupancy was short and was predicted to increase to a total of 8 hours per day with QID administration in approximately 75% of the population. Results of this study also suggest that longer duration of ³50% receptor occupancy following 5 mg BID administration can be achieved in approximately 75% of the population for 12 hours/day, which represents 67% (12 out of 18 hours) of waking time. These exploratory analyses were based on the relationship between plasma preladenant concentrations and receptor occupancy in healthy subjects. In the target population (Parkinson’s disease patients), the predicted receptor occupancy and duration may vary due to the inter-patient variability in PK and PD.
Based upon the established PK-PD relationship developed in this study it is quite probable that the A2A receptor occupancy can be maintained in a the majority of the subject population for the majority of the waking hours with preladenant doses around 5 mg BID and above. This additional analysis was helpful in estimating a lower dose (e.g., 5 mg) as a possible target for the minimal therapeutic effect in a real clinical trial scenario with a multiple dosing paradigm. Preliminary efficacy data in patients with moderate-to-severe Parkinson’s disease indicate that preladenant at doses of 5 and 10 mg BID significantly reduces off-time, which demonstrate a high accuracy of PET imaging biomarker approach to predict clinical efficacy. Thus, it was an effective demonstration of how such theoretical projections and associated dose-selection decisions can be made using pooled population Phase 1 PK data and actual PK-PD estimates from a single dose PET study. This obviated the need for conducting more extensive multiple dose PET studies and suggests that critical decision-making on clinical dose selection can be taken at an earlier stage.
This is the first study in which the relationship between preladenant plasma concentration and brain receptor occupancy has been determined using the A2A specific and selective radiotracer 11C-SCH442416. This study demonstrates the basic principles for determining proof-of-occupancy and proof-of-mechanism for a novel CNS target using characterization of PK-PD relationships, which enables Pharmaceutical companies to accelerate Go/No-Go decision-making in earlier clinical research and development.
4. Conclusions
The quantifiability of A2A receptor availability in the striatum and thalamus has been demonstrated in the 11C- SCH 442416 PET study [21]. A single oral dose of 50-200 mg preladenant achieved full A2A receptor blockade in the striatum. The receptor occupancy decreased in a dose and concentration-related manner from full blockade at 1-6 hours to 56-58% at 12 hours after 200 mg administration, and demonstrated dose- and time-dependent decreases as the dose was decreased to 10 mg preladenant and time increased from 1 to 6 or 12 hours respectively.
The plasma preladenant concentrations showed relationship with the A2A receptor occupancy, and this relationship was described by an Emax model. The Emax model-predicted plasma concentrations corresponding to 50%, 80%, and 90% specific receptor occupancy were 6 ng/mL, 25 ng/mL, and 50 ng/mL, respectively. Doses in the range of 5 to 100 mg were predicted to result in ≥80% receptor occupancy. Following a single 5 mg dose, the duration of occupancy was predicted to be short and increased with the dose and dosing frequency. A 50 mg dose, administered BID 8 hours apart, was estimated to result in ≥80% receptor occupancy in approximately 75% of the population for the entire duration of waking hours (18 hours/day). A 5 mg dose, administered BID, 8 hours apart, was estimated to provide ≥50% receptor occupancy in approximately 75% of the population for the majority of waking hours (12 hours/day).
Single doses of preladenant between 10 to 200 mg were well-tolerated. The Cmax and AUC values of preladenant increased in a dose-related manner. In this study we demonstrated the importance of PET imaging for establishing PK-PD relationships and utilizing this tool in confirming proof-of-target and dose guidance for Phase 2/3 clinical trials. This imaging technology is in line with the Critical Path Initiative – FDA’s national strategy for transforming the way FDA-regulated products are developed, evaluated, manufactured, and used.
Conflict of interest
ID Grachev: Full-time employee of Schering-Plough Research Institute at the time of the study conduct and does not hold any stocks or shares, or have other financial competing interests. DJ Brooks: Part-time employee of GE Healthcare at the time of the study conduct.
Acknowledgements
We gratefully acknowledge the staff of the Hammersmith PET Center, London, UK for successfully completing the PET scans; Imaging Research Solutions, Ltd (IRSL), Hammersmith Hospital, London, UK for the determination of specific binding potentials in the ROI, and the onsite manufacturing of 11C-SCH 442416; PPD Development, Richmond, Virginia, USA for assaying all plasma samples for preladenant, using an LC MS/MS assay; Hammersmith Medicines Research, Central Middlesex Hospital, London, UK for successful management of subjects enrollment, performing all required study procedures and safety monitoring. We particularly thank Safiye Osman for help with the development of the blood metabolite analysis method, Prof. Vincent J. Cunningham for help with the imaging data analysis, and Amol Tendolkar for help with PK-PD modeling. Drs. David Cutler, John Hunter and Steve Warrington receive our thanks for helpful discussions on the manuscript. This study was supported by Schering-Plough Research Institute, Kenilworth, NJ, USA.
References
Key References: 7, 18, 21, 34, 38
- Brooks DJ, Doder M. Depression in Parkinson’s disease. Curr Opin Neurol. 2001; 14: 465-470. [PubMed Abstract]
- Kuopio AM, Marttila RJ, Helenius H, Toivonen M, Rinne UK. The quality of life in Parkinson’s disease. Mov Disord. 2000; 15: 216-223. [CrossRef] [PubMed Abstract]
- Fredholm BB, Abbracchio MP, Burnstock G, et al. Nomenclature and classification of purinoceptors. Pharm Rev. 1994; 46: 143-156. [PubMed Abstract]
- Furlong TJ, Pierce KD, Selbie LA, Shine J. Molecular characterization of a human brain adenosine A2 receptor. Mol Brain Res. 1992; 15: 62-66. [CrossRef] [PubMed Abstract]
- Peterfreund RA, MacCollin M, Gusella J, Fink JS. Characterization and expression of the human A2a adenosine receptor gene. J Neurochem. 1996; 66: 362-368. [CrossRef] [PubMed Abstract]
- Wang W-F, Ishiwata K, Nonaka H, et al. Carbon-11-Labeled KF21213: A highly selective ligand for mapping CNS adenosine A2a receptors with positron emission tomography. Nucl Med Biol. 2000: 27; 541-546. [CrossRef] [PubMed Abstract]
- Schiffmann SN, Libert F, Vassart G, Vanderhaeghen J. Distribution of adenosine A2 receptor mRNA in the human brain. Neurosci Lett. 1991; 130; 177-181. [CrossRef] [PubMed Abstract]
- Svenningsson P, Le Moine C, Aubert I, Burbaud P, Fredholm BB, Bloch B. Cellular distribution of adenosine A2a receptor mRNA in the primate striatum. J Comp Neurol. 1998; 399: 229-240. [CrossRef] [PubMed Abstract]
- Mori A, Shindou T, Ichimura M, Nonaka H, Kase H. The role of adenosine A2a receptors in regulating GABAergic synaptic transmission in striatal medium spiny neurons. J Neurosci. 1998; 16: 605-611. [CrossRef]
- Ferrè S, Fredholm BB, Morelli M, Popoli P, Fuxe K. Adenosine dopamine receptor-receptor interactions as an integrative mechanism in the basal ganglia. Trends Neurosci. 1997; 20: 482-487. [CrossRef] [PubMed Abstract]
- Richardson PJ, Kase H, Jenner PG. Adenosine A2a receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol. Sci. 1997; 18: 338-344. [CrossRef] [PubMed Abstract]
- Kanda T, Shiozaki S, Shimada J, Suzuki F, Nakamura J. KF17837: A novel selective 2a receptor antagonist with anticataleptic activity. Eur J Pharmacol. 1994; 256: 263.[PubMed Abstract]
- Fenu S, Pinna A, Ongini E, Morelli M. Adenosine A2a receptor antagonism potentiates L-dopa-induced turning behavior and c-fos expression in 6-hydroxydopamine-lesioned rats. Eur J Pharmacol. 1997; 321: 143-147. [CrossRef] [PubMed Abstract]
- Mandhane SN, Chopde CT, Ghosh AK. Adenosine A2 receptors modulate haloperidol-induced catalepsy in rats. Eur J Pharmacol. 1997; 328: 135-141. [CrossRef] [PubMed Abstract]
- Kanda T, Jackson MJ, Smith LA, et al. Adenosine A2a antagonist: a novel antiparkinsonian agent that does not provoke dyskinesia in parkinsonian monkeys. Ann Neurol. 1998; 43: 507-513. [CrossRef] [PubMed Abstract]
- Grondin R, Bedard PJ, Tahar AH, Gregoire L Mori A, Kase H. Antiparkinsonian effect of a new selective adenosine A2a receptor antagonist in MPTP-treated monkeys. Neurology. 1999; 52: 1673-1677. [CrossRef] [PubMed Abstract]
- Kanda T, Jackson MJ, Smith LA, et al. Combined use of the adensoine A2a antagonist KW-6002 with L-DOPA or with selective D1 or D2 dopamine antagonists increases antiparkinsonian activity but not dyskinesia in MPTP-treated monkeys. Experimental Neurology. 2000; 162: 321-327. [PubMed Abstract]
- Moresco RM, Todde S, Belloli S, et al. In vivo imaging of adenosine A2A receptors in rat and primate brain using [11C]SCH442416. Eur J Nucl Med Mol Imaging. 2005; 32: 405-413. [CrossRef] [PubMed Abstract]
- Todde S, Moresco RM, Simonelli P, et al. Design, radiosynthesis, and biodistribution of a new potent and selective ligand for in vivo imaging of the adenosine A(2A) receptor system using positron emission tomography. J Med Chem. 2000; 43: 4359-4362. [PubMed Abstract]
- Mihara T, Noda A, Arai H, et al. Brain adenosine A2A receptor occupancy by a novel A1/A2A receptor antagonist, ASP5854, in rhesus monkeys: relationship to anticataleptic effect. J Nucl Med. 2008; 49: 1183-1188. [CrossRef] [PubMed Abstract]
- Grachev ID, Doder M, Brooks DJ, Hinz R. Quantitative in vivo Imaging of Adenosine A2A Receptors in the Human Brain Using 11C-SCH442416 PET: A Pilot Study. Journal of Diagnostic Imaging in Therapy. 2014; 1: 1-19. [CrossRef]
- Spinks TJ, Jones T, Bloomfield PM, et al. Physical characteristics of the ECAT EXACT3D positron tomograph. Phys Med Biol. 2000; 45: 2601-2618. [CrossRef] [PubMed Abstract]
- Ranicar AS, Williams CW, Schnorr L, et al. The on-line monitoring of continuously withdrawn arterial blood during PET studies using a single BGO/photomultiplier assembly and non-stick tubing. Med Prog Technol. 1991; 17: 259-264. [PubMed Abstract]
- Hammers A, Koepp MJ, Free SL, et al. Implementation and application of a brain template for multiple volumes of interest. Hum Brain Mapp. 2002; 15: 165-174. [CrossRef] [PubMed Abstract]
- Studholme C, Hill DL, Hawkes DJ. Automated three-dimensional registration of magnetic resonance and positron emission tomography brain images by multiresolution optimization of voxel similarity measures. Med Phys. 1997; 24: 25-35. [CrossRef] [PubMed Abstract]
- Robb RA, Barillot C. Interactive display and analysis of 3-D medical images. IEEE Trans Med Imag. 1989; 8: 217-226. [CrossRef] [PubMed Abstract]
- Robb RA, Hanson DP, Karwoski RA, Larson AG, Workman EL, Stacy MC. ANALYZE: a comprehensive, operator-interactive software package for multidimensional medical image display and analysis. Comput Med Imaging and Graph. 1989; 13: 433-454. [CrossRef] [PubMed Abstract]
- Cunningham VJ, Jones T. Spectral analysis of dynamic PET studies. J. Cereb. Blood Flow Metab. 1993; 13: 15-23. [CrossRef]
- Schmidt K. Which linear compartmental systems can be analyzed by spectral analysis of PET output data summed over all compartments? J Cereb Blood Flow Metab. 1999; 19: 560-569. [CrossRef] [PubMed Abstract]
- Gibaldi M, Perrier D. Chapter 11: Noncompartmental Analysis Based on Statistical Moment Theory, in: Pharmacokinetics. 2nd ed. New York: Marcel Dekker Inc., 409-417 (1982).
- Hardman J, Limbird L. Goodman and Gilman’s The Pharmacological Basis of Therapeutics. 9th ed. New York: McGraw-Hill, 37-41 (1996).
- Mishina M, Ishiwata K, Kimura Y, et al. Evaluation of distribution of adenosine A2A receptors in normal human brain measured with [11C]TMSX PET. Synapse. 2007; 61; 778-784. [CrossRef] [PubMed Abstract]
- Naganawa M, Kimura Y, Mishina M, et al. Quantification of adenosine A2A receptors in the human brain using [11C]TMSX and positron emission tomography. Eur J Nucl Med Mol Imaging. 2007; 34: 679-687. [CrossRef] [PubMed Abstract]
- Svenningsson P, Hall H, Sedvall G, Fredholm BB. Distribution of adenosine receptors in the postmortem human brain: An extended autoradiographic study. Synapse. 1997; 27: 322-335. [CrossRef] [PubMed Abstract]
- Tashiro M, Sakurada Y, Iwabuchi K, et al. Central effects of fexofenadine and cetirizine: measurement of psychomotor performance, subjective sleepiness, and brain histamine H1-receptor occupancy using 11C-doxepin positron emission tomography. J Clin Pharmacol. 2004; 44; 890-900. [PubMed Abstract]
- Petit-Taboué MC, Landeau B, Barré L, Onfroy MC, Noël MH, Baron JC. Parametric PET imaging of 5HT2A receptor distribution with 18F-setoperone in the normal human neocortex. J Nucl Med. 1999; 40; 25-32. [PubMed Abstract] [Reference Source]
- Hirani E, Gillies J, Karasawa A, et al. Evaluation of [4-O-methyl-11C]KW-6002 as a potential PET ligand for mapping central adenosine A2A receptors in rats. Synapse. 2001; 42: 164-176. [CrossRef] [PubMed Abstract]
- Brooks DJ, Papapetropoulos S, Vandenhende F, et al. An open-label, positron emission tomography study to assess adenosine A2A brain receptor occupancy of vipadenant (BIIB014) at steady-state levels in healthy male volunteers. Clin Neuropharmacol. 2010; 33: 55-60. [CrossRef] [PubMed Abstract]
Article information
Corresponding author: Igor D. Grachev.
Copyright: © 2014 Grachev ID et al.
How to cite: Grachev ID, Doder M, Brooks DJ, Hinz R. An in vivo positron emission tomography study of adenosine 2A receptor occupancy by preladenant using11C-SCH442416 in healthy subjects. J. Diagn. Imaging Ther. 2014; 1(1): 20-48 (https://dx.doi.org/10.17229/jdit.2014-0712-002).
Article history: Received 1 July 2014; Revised 8 July 2014; Accepted 9 July 2014; Published online 12 July 2014.
Archive link: JDIT-2014-0712-002
home »