In November and December 2025, the two largest clinical trials ever run with a glucagon-like peptide-1 receptor (GLP-1R) agonist in a neurodegenerative disease reported their results. Across the EVOKE and EVOKE+ trials, oral semaglutide did not slow cognitive or functional decline in 3,808 people with early Alzheimer’s disease, a finding that followed the Phase 3 Exenatide-PD3 trial’s null result in Parkinson’s disease earlier that year.
Both outcomes sit uneasily beside the positive Phase 2 LIXIPARK trial and a decade of preclinical and observational data pointing toward neuroprotection. This review sets out the molecular and central nervous system biology of the GLP-1 receptor, the mechanisms proposed to underlie its neuroprotective effects, and the clinical trial record in Parkinson’s and Alzheimer’s disease.
It then asks why well-powered trials have largely failed where smaller studies and real-world data succeeded, with attention to brain exposure, trial design, and patient selection, and outlines what the next generation of studies will need to test the hypothesis properly.
1. Introduction
The neuroprotection hypothesis for GLP-1 receptor agonists has just survived its hardest test, and it did not pass cleanly. On 24 November 2025, Novo Nordisk reported that oral semaglutide had failed to slow Alzheimer’s disease in the EVOKE and EVOKE+ trials, which together enrolled 3,808 participants and stand as the largest evaluation of this drug class in any neurodegenerative condition [1].
Ten months earlier, the Phase 3 Exenatide-PD3 trial had reached the same conclusion in Parkinson’s disease: no effect on progression across 194 patients followed for 96 weeks, with no treatment difference on dopamine-transporter imaging in the trial’s imaging substudy [2]. Yet in April 2024, the smaller LIXIPARK trial had shown the opposite, with lixisenatide slowing motor decline in early Parkinson’s disease over 12 months [3].
That contradiction is the real state of the field, and it is more instructive than either the optimism of the preclinical literature or the gloom of the recent headlines. A receptor first understood as a pancreatic switch for insulin turned out to sit across circuits that govern reward, memory, mood, and movement, and the drugs that engage it have become some of the most-studied molecules in medicine.
The question is no longer whether GLP-1 receptor activation can protect neurons in a model system. It can, and has, in study after study. The question is whether that effect survives the move into human disease. This review covers the molecular identity and central nervous system distribution of the GLP-1 receptor, the mechanisms proposed to underlie its neuroprotective effects, and the clinical trial record in Parkinson’s and Alzheimer’s disease. It argues that the failures of EVOKE and Exenatide-PD3 narrow the hypothesis rather than refute it, pointing toward brain-penetrant compounds, biomarker-anchored endpoints, and patients selected by biology rather than diagnosis.
2. The GLP-1 receptor: from gut hormone to brain target
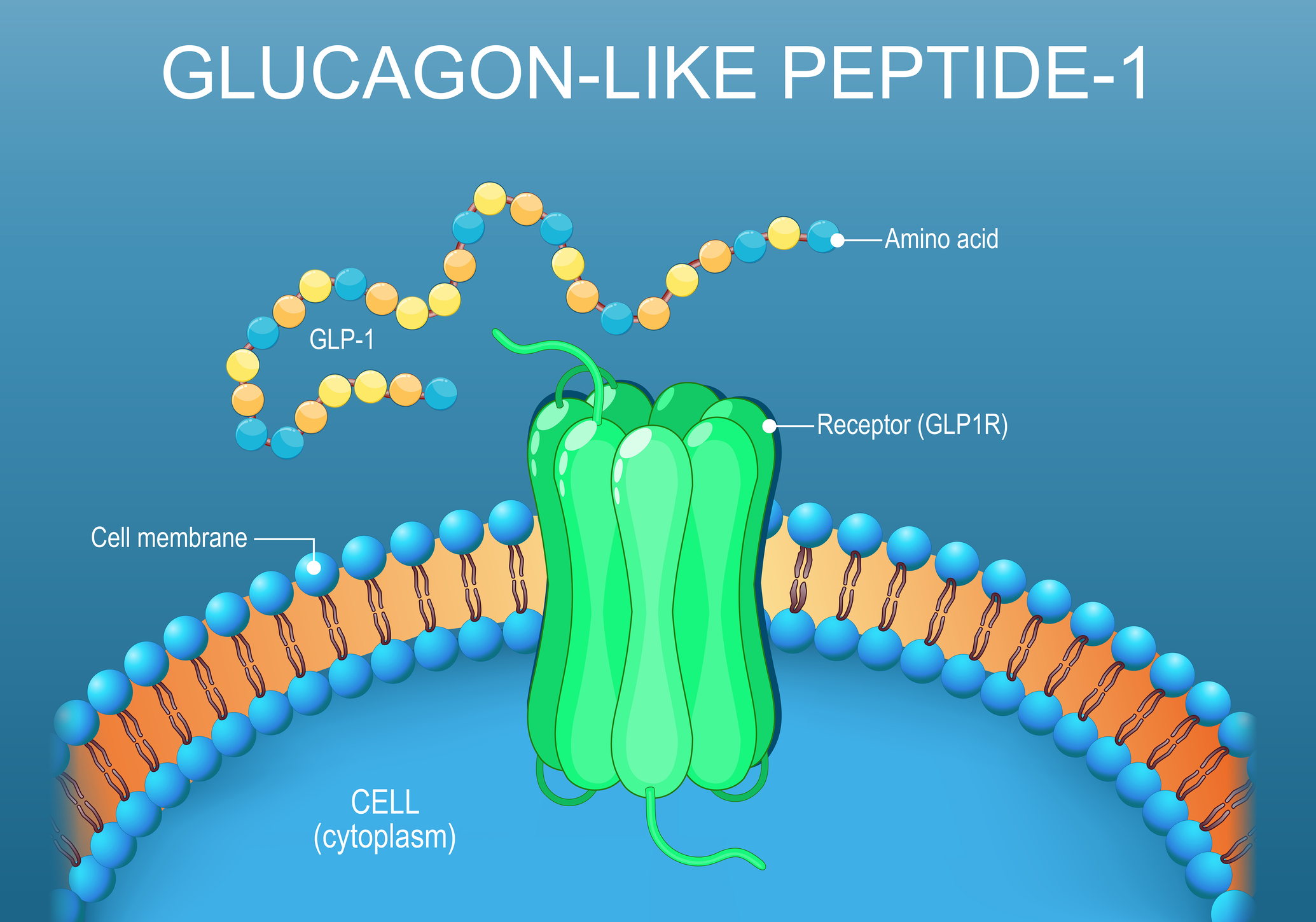
Glucagon-like peptide-1 is a 30 to 31-amino acid incretin hormone cleaved from proglucagon. It is released by intestinal L-cells after a meal and, separately, by preproglucagon neurons in the nucleus tractus solitarius of the brainstem, which gives the peptide both a peripheral endocrine role and a central neural one. Its receptor, GLP-1R, is a class B G-protein-coupled receptor. Activation couples to Gs, raises intracellular cyclic AMP, and engages protein kinase A. In the pancreas, this amplifies glucose-dependent insulin secretion and suppresses glucagon; in the gut and brain, it slows gastric emptying and reduces appetite.
A nomenclature note matters here because it is a common source of confusion. The proglucagon precursor yields two distinct peptides with two distinct receptors: GLP-1 acting at GLP-1R, and GLP-2 acting at GLP-2R, the latter largely confined to intestinal growth and repair. There is no established “GLP-3” peptide or receptor in human physiology. Compounds marketed as dual or triple agonists, such as tirzepatide, act at combinations of the GLP-1, GIP, and glucagon receptors rather than at any third GLP receptor.
When exenatide received FDA approval in 2005, GLP-1R was understood primarily as a pancreatic target, and the incretin system looked like a clean metabolic story. Liraglutide, lixisenatide, dulaglutide, and semaglutide followed, and the class went on to become one of the most commercially successful in pharmaceutical history, later expanding into obesity.
The brain was an afterthought. It should not have been. GLP-1 receptors are expressed in the hypothalamus, hippocampus, cortex, amygdala, ventral tegmental area, nucleus accumbens, and lateral septum, with the highest density in the lateral septum. That distribution overlaps almost exactly with the circuits implicated in neurodegeneration, addiction, and mood disorders, which is why neuroscientists began paying attention.
3. Mechanisms proposed to underlie neuroprotection
The preclinical literature is now large enough to describe the proposed mechanisms with reasonable confidence, even where the human relevance of each remains open. They overlap rather than compete.
Anti-inflammatory and glial effects
In preclinical models, GLP-1R activation suppresses microglial activation and lowers central production of TNF-alpha, IL-1 beta, and IL-6. The most cited single result is from Yun and colleagues in 2018, who showed that NLY01, a long-acting PEGylated GLP-1R agonist, blocked the microglia-driven conversion of resting astrocytes into the neurotoxic A1 phenotype in mouse models, a process implicated in neuronal death in both Parkinson’s and Alzheimer’s disease [4]. This work has been widely read as evidence that neuroinflammation is a driver of neurodegeneration rather than merely a downstream marker, although that interpretation remains the subject of active investigation.
Neurogenesis, synaptic plasticity, and metabolic support
GLP-1R stimulation promotes hippocampal neurogenesis and enhances synaptic plasticity, partly through brain-derived neurotrophic factor and modulation of glutamatergic signalling. In a foundational 2009 study, Li and colleagues found that GLP-1R stimulation preserved both cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism [5].
A parallel line of work frames Alzheimer’s disease as a state of central insulin resistance, sometimes called type 3 diabetes, in which GLP-1R agonism restores neuronal insulin signalling, improves mitochondrial function, and reduces oxidative stress. The breadth of these effects is the point: the receptor does not appear to act through a single disease-specific mechanism but through a general increase in neuronal resilience.
Reward circuitry
One of the less expected findings concerns reward. Functional imaging studies show that GLP-1R agonists blunt neural responses to reward cues in the ventral tegmental area and nucleus accumbens, and retrospective and early prospective data link their use to reduced intake of alcohol and nicotine. This places the receptor at the intersection of metabolic, neurological, and psychiatric research, and is the basis for the addiction work discussed below.
4. Clinical trials in Parkinson’s disease
Parkinson’s disease is where the clinical story is most developed, and where the gap between phases is most visible.
The first encouraging human signal came from Athauda and colleagues in 2017. In a Phase 2 trial of 62 patients with moderate Parkinson’s disease, once-weekly exenatide produced a motor advantage in the off-medication state that persisted twelve weeks after the drug was stopped, which the authors interpreted as a possible signal of disease modification rather than symptomatic relief [6]. LIXIPARK, published in the New England Journal of Medicine in 2024, was the strongest result to date.
It randomised 156 participants with early disease to daily subcutaneous lixisenatide or placebo for 12 months, followed by a 2-month washout. At 12 months, the on-medication MDS-UPDRS Part III score was 14.9 in the lixisenatide group versus 18.8 in the placebo group, a between-group difference of 3.08 points (P = 0.007) [3].
The effect held after washout, with off-medication scores of 17.7 versus 20.6. Two caveats temper the result: nausea occurred in 46% of lixisenatide-treated participants and vomiting in 13%, raising the possibility of functional unblinding, and the MDS-UPDRS is a clinical scale rather than a direct measure of dopaminergic neuron survival.
Then came the reckoning. Exenatide-PD3, reported in The Lancet in early 2025, was the largest and longest trial of a GLP-1R agonist in Parkinson’s disease: 194 patients, once-weekly exenatide, 96 weeks. It found no evidence that exenatide slowed progression, and dopamine-transporter SPECT imaging in the trial’s substudy showed no treatment difference [2].
In a statement issued by University College London, the trial’s senior investigator, Thomas Foltynie, characterised the negative result as a major disappointment for patients with Parkinson’s disease and for the research community. A Phase 2 trial of liraglutide told a related story, with no clear motor benefit but possible gains in non-motor symptoms and quality of life.
Pooled analyses of the smaller trials conflict rather than converge. The most recent and methodologically rigorous of them, restricted to four placebo-controlled trials in 667 patients, found no significant difference from placebo on MDS-UPDRS Part III in either the on- or off-medication state [7]. Earlier meta-analyses did report a small on-medication motor benefit, roughly one to three points, but that effect is heterogeneous and fragile: in at least one pooled analysis, it lost statistical significance once the lixisenatide trial was removed [8]. Those positive estimates also rest largely on the early-phase studies, and the single most heavily weighted Phase 3 result, the null Exenatide-PD3 trial, argues against a robust class-wide effect. Taken together, the evidence suggests that class-wide disease modification in Parkinson’s, at the compounds and doses tested so far, is unlikely, even if a responder subgroup may yet exist.
Table 1. Major randomised trials of GLP-1 receptor agonists in neurodegenerative disease.
PD, Parkinson’s disease; AD, Alzheimer’s disease; UPDRS-III / CDR-SB, clinical rating scales; DaT-SPECT, dopamine-transporter imaging.
5. Clinical trials in Alzheimer’s disease
Alzheimer’s disease is where the 2025 data forced the largest revision, and where the contrast between observational promise and trial outcome is sharpest.
Real-world and observational evidence have been consistently encouraging. Target-trial-emulation and cohort analyses report that diabetic patients on GLP-1R agonists develop dementia and Alzheimer’s disease at lower rates than those on comparator antidiabetic drugs, and pooled analyses of cardiovascular-outcome trials have pointed in the same direction [9]. These are hypothesis-generating signals, vulnerable to confounding by indication and healthy-user effects, but they were strong enough to justify large prospective trials.
Those trials have now been reported, and they complicate the picture. The ELAD Phase 2b trial of liraglutide, published in Nature Medicine, randomised 204 patients with mild to moderate disease to daily liraglutide or placebo for 52 weeks. Its primary endpoint, cerebral glucose metabolism, was negative (difference -0.17, P = 0.14). However, the secondary and exploratory endpoints were striking: roughly 18% slower decline on a cognitive composite and close to 50% less loss of grey-matter volume on MRI [10]. ELAD was not powered for those outcomes, so it reads as encouraging but inconclusive. EVOKE and EVOKE+ were neither.
These twin Phase 3 trials enrolled 3,808 participants with early symptomatic Alzheimer’s disease confirmed by amyloid biomarkers and tested once-daily oral semaglutide against placebo. Neither showed any effect on the primary endpoint, the Clinical Dementia Rating-Sum of Boxes at 104 weeks, nor on secondary cognitive and functional scales, and a pooled analysis found no delay in progression from mild cognitive impairment to dementia (hazard ratio 0.96) [1]. Semaglutide lowered several cerebrospinal fluid Alzheimer’s markers by up to about 10% and produced the expected weight loss, but none of this translated into clinical benefit; Novo Nordisk subsequently discontinued the one-year extension phase [1,11].
The pattern is uncomfortable but clear: the single largest, best-powered Alzheimer’s trial of this class was negative, while the positive evidence is observational or confined to secondary endpoints in underpowered studies. A biomarker effect without a clinical one, as seen in EVOKE, is exactly the dissociation that should make researchers cautious about surrogate measures.
6. The brain-access hypothesis and why it is contested
One influential hypothesis for making sense of these mixed results, advanced by Christian Holscher, proposes that neuroprotective efficacy tracks a compound’s ability to reach the brain. On this view, exenatide and lixisenatide cross the blood-brain barrier readily and showed positive effects in Parkinson’s disease; liraglutide crosses poorly and showed limited effects; and the PEGylated agonist NLY01, which on this account does not effectively reach the brain despite being engineered for penetrance, was negative in a Phase 2 Parkinson’s trial [12,13]. The EVOKE result is consistent with this logic: semaglutide is a large, acylated molecule with poor brain penetrance, and it did not show benefit in Alzheimer’s disease. In Holscher’s reading, that is the expected outcome, not a surprise.
The hypothesis is attractive, and the EVOKE data strengthen it, but it is not settled, and two problems deserve attention. First, exenatide crosses well and still fails in the Phase 3 Exenatide-PD3 trial, so brain access cannot be the whole story. Second, and more fundamentally, it is not clear that these drugs cross the blood-brain barrier in the classical sense at all.
Individual speakers at a 2024 National Academies workshop suggested that GLP-1R agonists may reach the brain mainly through specialised uptake at the circumventricular organs, regions that sit outside the standard barrier, rather than by penetrating it; the proceedings note that these summarised points were not formally endorsed by the National Academies [14]. If this proves correct, “blood-brain barrier penetrance” may be the wrong language for a real but poorly understood phenomenon, and compound design should target whatever route actually delivers these peptides to the relevant neurons.
Either way, the practical implication is similar, and it is arguably the most consequential consideration for anyone planning future work: on the brain-access hypothesis, novel dual GLP-1/GIP agonists engineered specifically for brain entry would be more rational next candidates to test than repurposed diabetes drugs chosen for market availability. This remains a hypothesis to be tested rather than an established conclusion.
7. Signals in depression and addiction
The central nervous system applications extend past movement and memory disorders, though the evidence thins as the indication moves away from Parkinson’s. For depression, a meta-analysis of randomised and cohort data reported a statistically significant reduction in depression scores among GLP-1R agonist users relative to controls [15].
The addiction signal is the most novel. Across animal models, GLP-1R agonists reduce reward-seeking behaviour for several substance classes, and human observational data are beginning to align; a 2025 review concluded that GLP-1R agonists reduce alcohol intake, lower the motivation to drink, and prevent relapse drinking in preclinical models [16].
If validated in adequately powered human trials, this would be a genuinely new mechanism for substance use disorders, rooted in metabolic signalling rather than monoamine modulation. For now, it remains the earliest-stage part of the field.
8. Regulatory status and translational considerations
No GLP-1 receptor agonist is approved for any neurological or psychiatric indication. The approved uses of this class are type 2 diabetes, chronic weight management, and, for several agents, reduction of cardiovascular risk. Every neuroprotective application discussed here is investigational or off-label. We are aware of no breakthrough or accelerated regulatory designation for a neurodegenerative indication in this class, and the recent Phase 3 failures are likely to make near-term regulatory filings in this space unlikely. That status should frame how the evidence is read and communicated.
The harder lessons are methodological, and they shape what the next round of trials should look like. Four points stand out. Compound selection should be driven by demonstrated brain exposure rather than commercial prominence; the agents that engage central targets, not the best-selling ones, are the ones worth testing, and brain-penetrant next-generation designs should take priority. Endpoints need to move beyond clinical rating scales. The MDS-UPDRS and CDR-SB cannot cleanly separate symptomatic relief from disease modification, and EVOKE showed the converse failure mode, biomarker movement without clinical benefit, so future trials should anchor on objective measures such as dopamine-transporter SPECT, neurofilament light chain, and tau, amyloid, and neuroinflammation PET. Blinding has to be protected; the gastrointestinal side effects and visible weight loss typical of this class threaten the integrity of trials with subjective outcomes, which again argues for objective endpoints.
Patient stratification probably matters more than any single drug choice, because the variable results likely conceal responder subgroups; stratifying by metabolic status, by genetic risk such as GBA and LRRK2 variants in Parkinson’s or APOE in Alzheimer’s, by disease stage, and potentially by GLP-1R polymorphism is the most plausible route to a positive trial.
A final point concerns reproducibility rather than design. Some of the inconsistency across preclinical reports reflects differences in the compounds, doses, and assay conditions used, so standardised reporting of peptide characterisation and experimental conditions would make the literature easier to interpret and to replicate.
9. Imaging and biomarker endpoints
The sharpest biomarker lesson from this literature comes from a negative trial. In EVOKE and EVOKE+, oral semaglutide lowered several cerebrospinal-fluid markers of Alzheimer’s pathology, including phosphorylated tau and total tau, by up to roughly 10%, yet produced no measurable slowing of clinical decline; plasma neurofilament light chain and glial fibrillary acidic protein, the fluid markers that track neuroaxonal injury and astrocytic activation, did not improve and in some analyses drifted slightly upward [1]. A drug can move a fluid biomarker in the apparently favourable direction and still fail the patient. That dissociation is the strongest practical argument for treating cerebrospinal fluid and plasma markers in these trials as exploratory readouts of target engagement rather than as stand-ins for benefit.
Imaging endpoints carry the same caution in a different form. The dopamine-transporter SPECT substudy embedded in Exenatide-PD3 showed no separation between exenatide and placebo, mirroring the null clinical result and offering no imaging signal of preserved nigrostriatal terminals [2]. On the Alzheimer’s side, amyloid PET and plasma p-tau217 now do real work at the front door of trials, confirming that enrolled participants actually carry the pathology in question, as the amyloid-confirmed EVOKE cohorts illustrate. Tau PET is the more informative progression marker because it maps where pathology sits and tracks cognitive change more closely than amyloid load does. No GLP-1 trial has yet reported a positive treatment effect on any of these imaging measures, which is itself worth stating plainly.
Structural MRI gives the most encouraging-looking number in the whole class, and it needs the most careful handling. ELAD reported roughly 50% less grey-matter volume loss with liraglutide than with placebo [10]. Read in isolation, that sounds like neuroprotection. Read in context, it is a secondary, exploratory outcome from a Phase 2 trial whose primary endpoint, change in cerebral glucose metabolism, was negative, and whose cognitive signal was modest. The volumetric finding is a hypothesis worth testing in a powered trial with volume change as a prespecified endpoint, not a demonstration that the drug protects brain tissue.
The way through is to assign each measure an explicit job before the trial starts rather than after the results arrive. Amyloid PET and plasma p-tau confirm eligibility. Cerebrospinal fluid and plasma analytes index target engagement and pharmacodynamics. Tau PET and volumetric MRI are candidate markers of disease trajectory that have not been qualified as surrogates in this drug class. Neurofilament light chain sits slightly apart, useful both as a marker of ongoing neuroaxonal damage and as a safety signal when it moves the wrong way, as it arguably did in EVOKE. None of these endpoints has been validated against clinical outcomes for GLP-1R agonists specifically, and EVOKE is the cautionary case of how readily a biomarker shift and a clinical result can be conflated. Until that qualification work is done, the imaging and fluid measures are instruments for understanding the mechanism and selecting patients, while the question of whether these drugs modify the disease still belongs to the clinical endpoints.
10. Conclusion
The question has changed. For fifteen years, it was whether GLP-1 receptor activation could protect neurons, and the preclinical answer was a repeated yes. The 2025 trials asked something harder: whether the drugs already on the market, at the doses and in the populations studied, can slow human neurodegeneration, and for semaglutide in Alzheimer’s disease and exenatide in Parkinson’s disease, the answer was no.
What survives that test is narrower and more testable than the broad claim it replaces: that brain-penetrant agonism, delivered early, in patients chosen by their biology rather than their diagnosis, and judged against biomarkers rather than questionnaires, might still work.
These drugs reached patients with diabetes long before the science was ready to ask them to protect the brain. Whether they ever will depends less on the receptor, whose biology is no longer in serious doubt, than on two engineering problems that the recent failures have brought into focus: getting the compound to the right neurons, and getting the right patients into the trial. The next generation of studies is, in that sense, as much a test of trial design and molecular delivery as of the hypothesis itself.
References
- Meissner WG, Remy P, Giordana C, et al. LIXIPARK Study Group. Trial of lixisenatide in early Parkinson’s disease. N Engl J Med. 2024 Apr 4;390(13):1176-1185. doi: 10.1056/NEJMoa2312323.
- Athauda D, Maclagan K, Skene SS, et al. Exenatide once weekly versus placebo in Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet. 2017 Oct 7;390(10103):1664-1675. doi: 10.1016/S0140-6736(17)31585-4.
- Vijiaratnam N, Girges C, Auld G, et al. Exenatide once a week versus placebo as a potential disease-modifying treatment for people with Parkinson’s disease in the UK: a phase 3, multicentre, double-blind, parallel-group, randomised, placebo-controlled trial. Lancet. 2025 Feb 22;405(10479):627-636. doi: 10.1016/S0140-6736(24)02808-3.
- Cummings JL, Atri A, Sano M, et al. Efficacy and safety of oral semaglutide 14 mg (flexible dose) in early-stage symptomatic Alzheimer’s disease (evoke and evoke+): two phase 3, randomised, placebo-controlled trials. Lancet. 2026 May 30;407(10544):2167-2179. doi: 10.1016/S0140-6736(26)00459-9.
- Edison P, Femminella GD, Ritchie C, et al. Liraglutide in mild to moderate Alzheimer’s disease: a phase 2b clinical trial. Nat Med. 2026 Jan;32(1):353-361. doi: 10.1038/s41591-025-04106-7.
- Li Y, Perry T, Kindy MS, et al. GLP-1 receptor stimulation preserves primary cortical and dopaminergic neurons in cellular and rodent models of stroke and Parkinsonism. Proc Natl Acad Sci U S A. 2009 Jan 27;106(4):1285-90. doi: 10.1073/pnas.0806720106.
- Yun SP, Kam TI, Panicker N, et al. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018 Jul;24(7):931-938. doi: 10.1038/s41591-018-0051-5.
- Holscher C. The neuroprotective properties of GLP‐1R agonists correlate with their ability to cross the blood‐brain barrier. Alzheimers Dement. 2025 Jan 9;20(Suppl 6):e088069. doi: 10.1002/alz.088069.
- Stefanou MI, Panagiotopoulos E, Tentolouris A, et al. Efficacy and safety of glucagon-like peptide-1 receptor agonists in Parkinson’s disease: a systematic review and meta-analysis of randomized placebo-controlled clinical trials. Ther Adv Neurol Disord. 2026 Jan 31;19:17562864251408269. doi: 10.1177/17562864251408269.
- Chen X, Zhao P, Wang W, Guo L, Pan Q. The antidepressant effects of GLP-1 receptor agonists: a systematic review and meta-analysis. Am J Geriatr Psychiatry. 2024 Jan;32(1):117-127. doi: 10.1016/j.jagp.2023.08.010.
- Jerlhag E. GLP-1 receptor agonists: promising therapeutic targets for alcohol use disorder. Endocrinology. 2025 Feb 27;166(4):bqaf028. doi: 10.1210/endocr/bqaf028.
- Zhang P, Mao C, Sun A, et al. Real-world observations of GLP-1 receptor agonists and SGLT-2 inhibitors as potential treatments for Alzheimer’s disease. Alzheimers Dement. 2025 Sep;21(9):e70639. doi: 10.1002/alz.70639.
- National Academies of Sciences, Engineering, and Medicine; Health and Medicine Division; Board on Health Sciences Policy; Forum on Neuroscience and Nervous System Disorders. Examining Glucagon-Like Peptide-1 Receptor (GLP-1R) Agonists for Central Nervous System Disorders: Proceedings of a Workshop. Norris SMP, Childers E, Pool R, editors. Washington (DC): National Academies Press (US); 2025 May 26. PMID: 40029954.
- McGarry A, Rosanbalm S, Leinonen M, et al. Safety, tolerability, and efficacy of NLY01 in early untreated Parkinson’s disease: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2024 Jan;23(1):37-45. doi: 10.1016/S1474-4422(23)00378-2.
- Novo Nordisk A/S. Novo Nordisk announces topline results from the evoke and evoke+ trials of oral semaglutide in early-stage symptomatic Alzheimer’s disease. Company announcement, 24 November 2025.
- Helal MM, AbouShawareb H, Abbas OH, et al. GLP-1 receptor agonists in Parkinson’s disease: an updated comprehensive systematic review with meta-analysis. Diabetol Metab Syndr. 2025 Aug 23;17(1):352. doi: 10.1186/s13098-025-01888-1.
Disclaimer: The information presented in this article is intended for educational, scientific, and informational purposes only. It does not constitute medical advice, diagnosis, treatment recommendations, regulatory guidance, or professional healthcare consultation. Readers should not rely on the content of this article as a substitute for advice from qualified healthcare professionals.
The discussion of glucagon-like peptide-1 receptor (GLP-1R) agonists in neurological and psychiatric disorders reflects the current scientific literature and clinical evidence available at the time of publication. Many of the neuroprotective, neurodegenerative, psychiatric, and addiction-related applications discussed remain investigational, and no GLP-1 receptor agonist is currently approved by regulatory authorities for the treatment or prevention of Alzheimer’s disease, Parkinson’s disease, depression, substance use disorders, or other neurological indications unless specifically stated otherwise.
While every effort has been made to ensure the accuracy of the information presented, Open MedScience, the authors, editors, reviewers, and contributors make no representations or warranties regarding the completeness, accuracy, reliability, or currency of the content. Scientific understanding, clinical evidence, regulatory positions, and treatment recommendations may change as new research becomes available.
References to specific drugs, clinical trials, companies, institutions, or products are provided solely for scientific discussion and do not imply endorsement, recommendation, or commercial promotion. Readers are encouraged to consult original publications, regulatory documentation, and qualified healthcare professionals before making decisions related to patient care, research, or healthcare policy.
Open MedScience accepts no liability for any loss, injury, damage, or consequences arising from the use of, or reliance upon, the information contained in this article.