
Modern Content Systems in Medical Affair
 By
By
Understand the role of Modern Content Systems in streamlining medical content across diverse digital channels effectively.
By
Understand the role of Modern Content Systems in streamlining medical content across diverse digital channels effectively.
 By
By
Learn how to get ready for a healthcare compliance audit with structured processes and continuous monitoring for better outcomes.
 By
By
Resources for successful lab operations enhance efficiency through effective software, skilled staff, reliable equipment, and compliance. Image for illustration only. People depicted are models.
 By
By
Medical Device Regulation ensures safety, enhances standards, fosters innovation, and supports effective healthcare delivery.
 By
By
The regulations in 21 CFR Part 212 ensure PET drugs are manufactured under strict quality control and safety standards.
 By
By
Good Manufacturing Practices ensure the consistent quality, safety, and efficacy of pharmaceutical products through rigorous production standards.
 By
By
ICH Q10 is a global model for managing pharmaceutical quality, ensuring compliance, continuous improvement, and risk manage.
 By
By
Q9(R1) Quality Risk Management provides a comprehensive framework for assessing, controlling, and mitigating pharmaceutical quality risks.
 By
By
ICH Guideline E8 establishes essential principles for designing, conducting, and reporting scientifically valid and ethically sound clinical trials.
 By
By
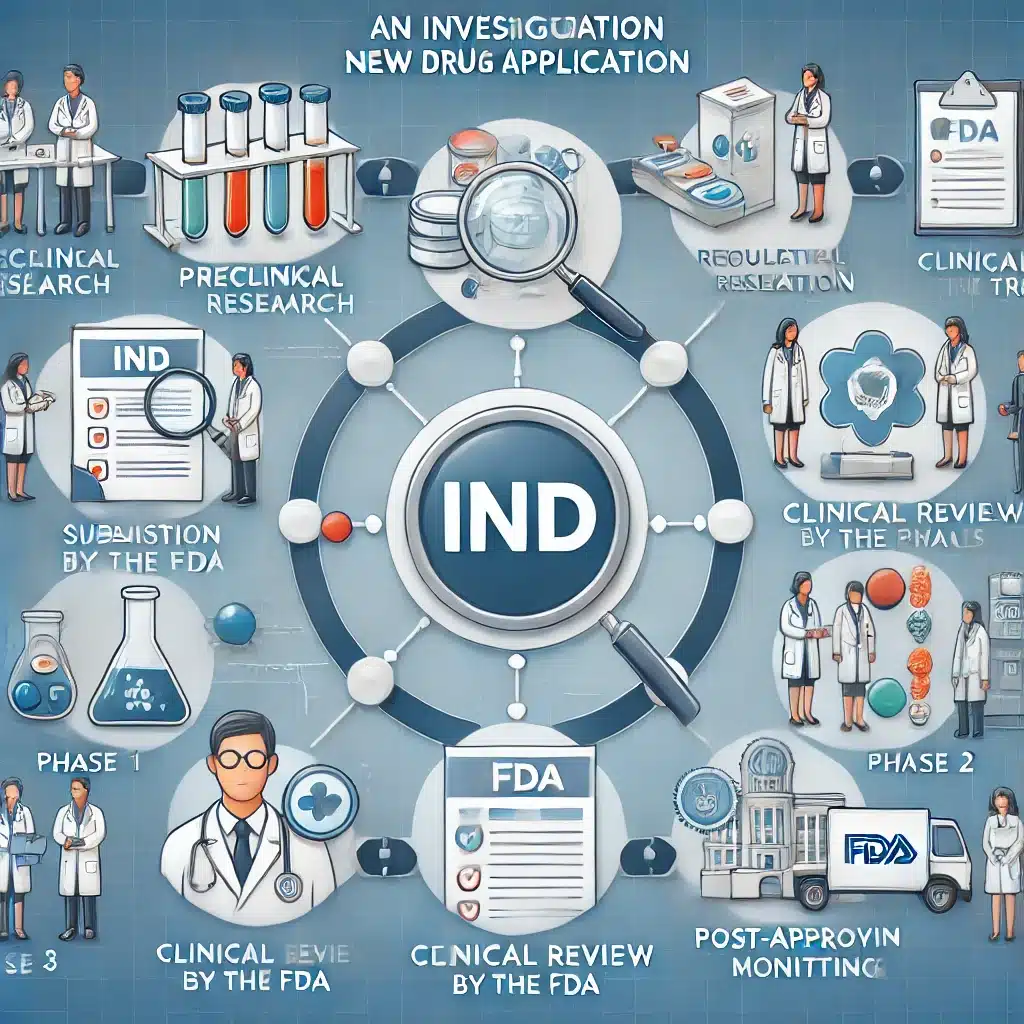
The IND application allows researchers to test investigational drugs in humans, ensuring safety and regulatory compliance throughout development.
 By
By
Radioactivity measurement ensures accurate detection and quantification of radiation levels for safety and regulatory compliance.
 By
By
The ICH Q7 guideline ensures consistent manufacturing practices, maintaining API quality, purity, and safety throughout production processes.
 By
By
Regulatory radiopharmaceutical production requires strict adherence to safety standards, ensuring quality and patient safety.
 By
By
Ensuring compliance with EU MDR poses significant challenges for medical device manufacturers, including increased scrutiny, financial strain, and data protection demands. Image for illustration only. People depicted are models.
 By
By
Adhering to environmental regulations in radiopharmaceutical production ensures safety, reduces waste, and protects the surrounding ecosystem.
 By
By
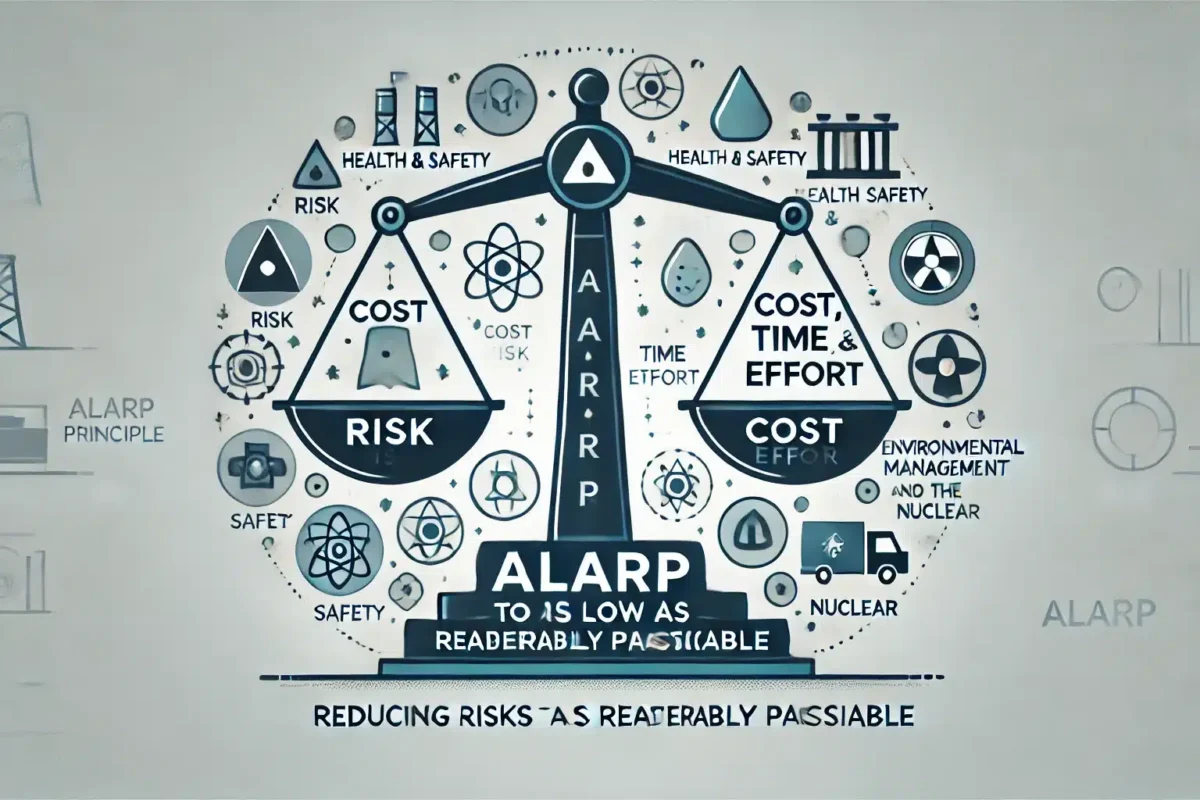
The ALARP principle ensures risks are reduced to the lowest practicable level, balancing cost, time, and safety measures.
 By
By
The Ionising Radiations Regulations 2017 mandate strict controls to protect workers and the public from ionising radiation risks.
 By
By
Medical Device Regulatory Compliance ensures safety and effectiveness through stringent testing and approval processes globally.