Molecular Imaging Technology: Use of PET in Clinical Microdose Studies
 By
By
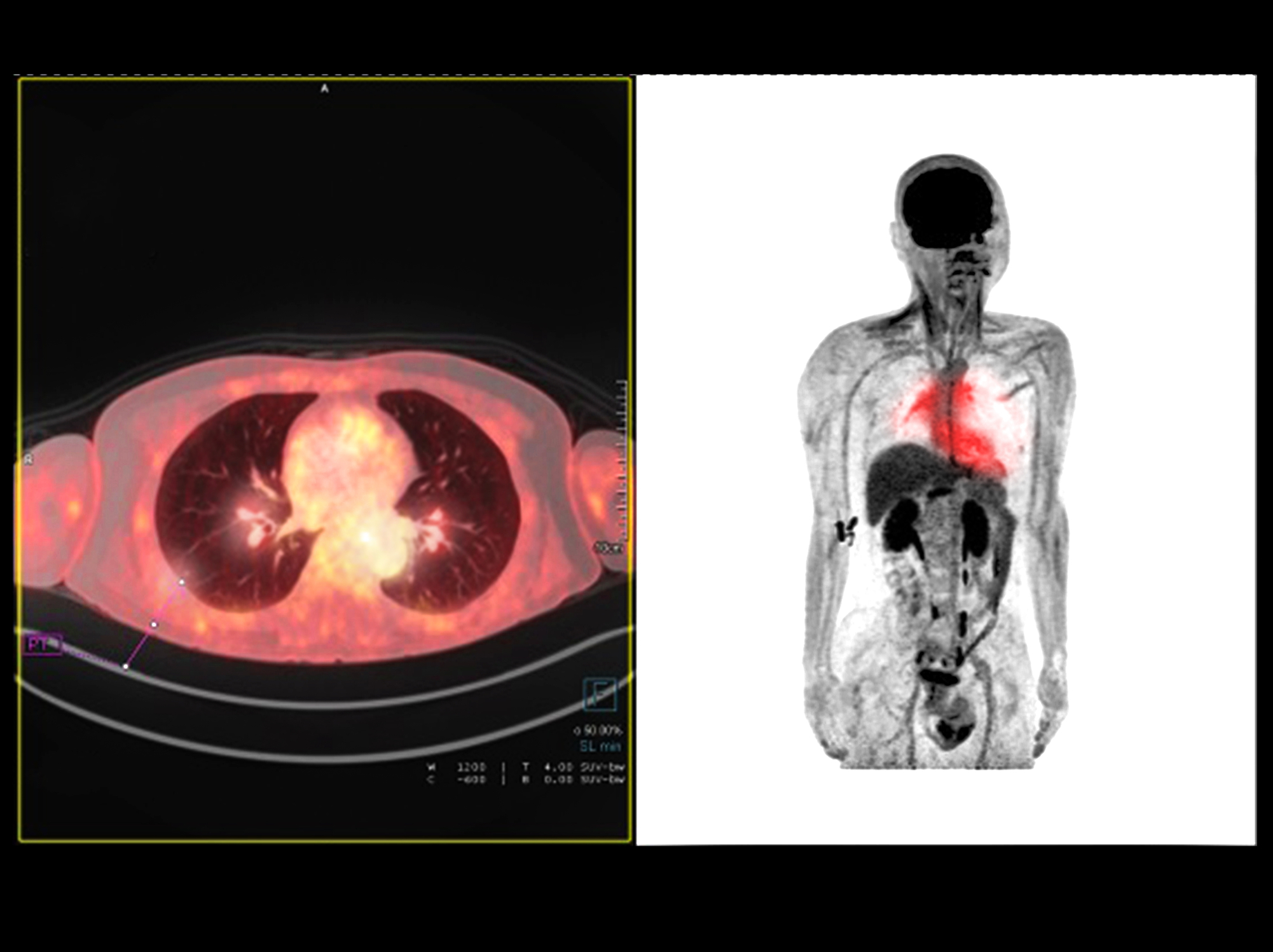
PET imaging is used in oncology, neurology and cardiology.
Microdose studies were first introduced in 2004 as a robust tool to assist in the drug development process. The idea is to obtain the pharmacokinetic profile of a drug in plasma from human volunteers rapidly and cost-effectively using a sub-therapeutic microdose (≤100 μg). However, in 2006, the US Food and Drug Administration (FDA) allowed pharmaceutical companies to conduct microdose research for the early drug applicant selection of the lead drug compound.
The drug is subjected to ultrasensitive analytical methods such as accelerator mass spectrometry (AMS) and positron emission tomography imaging. Microdosing became a section of the exploratory IND (investigational new drug) approach to increase the efficiency of the drug development process.
Furthermore, toxicology testing is mandatory by the regulatory authorities before microdose studies can be conducted in humans. This requires the drug applicant to have at least a reasonable pharmacokinetic profile and be selected at the earliest stage of drug development. In 2008, Japan’s regulatory authorities recommended microdosing in clinical trials.
A precondition for extrapolating plasma or cell concentrations of a drug – after administration of a microdose – must achieve a therapeutic dosage where plasma and tissue increase linearly as increasing medication doses as administered. Microdose research has demonstrated dose-independent pharmacokinetics for several drugs.
However, the query of dose linearity continues to be considered the main drawback to the microdosing idea. In addition, pharmacokinetic versions for predicting therapeutic medication behaviour in human microdose data must be validated to ensure that microdosing can fulfil its guarantee to shorten the drug advancement process.
home » microdosing
PET imaging is used in oncology, neurology and cardiology.