
REVIEW ARTICLE
Simon G. Patching
School of BioMedical Sciences and the Astbury Centre for Structural Molecular Biology, University of Leeds, Leeds, LS2 9JT, UK
Table of Contents
Abstract
Nuclear magnetic resonance (NMR) spectroscopy is a principal well-established technique for analysis of chemical, biological, food and environmental samples. This article provides an overview of the properties and applications of NMR active nuclei (39 nuclei of 33 different elements) used in NMR measurements (solution- and solid-state NMR, magnetic resonance spectroscopy, magnetic resonance imaging) with biological and biomedical systems and samples. The samples include biofluids, cells, tissues, organs or whole body from different organisms (humans, animals, bacteria, fungi, plants) for detecting and quantifying metabolites or environmental samples (water, soils, sediments). Isolated biomolecules (peptides, proteins, nucleic acids) can be analysed for elucidation of atomic-resolution structure, conformation and dynamics and for characterisation of ligand and drug binding, and of protein-ligand, protein-protein and protein-nucleic acid interactions. NMR can be used for drug screening and pharmacokinetics and to provide information in the design and discovery of new drugs. NMR can also measure translocation of ions and small molecules across lipid bilayers and membranes, characterise structure, phase behaviour and dynamics of membranes and elucidate atomic-resolution structure, orientation and dynamics of membrane-embedded peptides and proteins.
Keywords: biological and biomedical applications; drug screening; dynamics; magnetic resonance imaging; membrane proteins; metabolomics; MRI; NMR active nuclei; nuclear magnetic resonance; protein structure
1. Introduction
Nuclear magnetic resonance (NMR) spectroscopy is one of the principal techniques used for analysis of biological and biomedical systems and samples. This can include the identification, quantification and monitoring of ions, small molecules and biomolecules in studies of metabolism and biological function in human and animal cells and tissues, bacterial cells and spores, fungi and plants. Similar types of measurements can be performed on environmental samples such as water, soils and sediment. NMR is able to elucidate atomic-resolution structure, conformation, molecular mechanism, dynamics and exchange processes (on timescales of picoseconds to seconds) in biomolecules, especially peptides, proteins and nucleic acids. NMR can be used for the observation, quantification and characterisation of ligand and drug binding to biomolecules, and for characterisation of ligand-protein, protein-protein and protein-nucleic acid interactions. NMR can be used for drug screening and it can acquire structural, binding and kinetic information for the design and discovery of new drugs. It can then monitor the absorption, distribution, metabolism and excretion (ADME) of administered drugs in pharmacokinetics studies. NMR can be used for the observation, quantification and kinetic characterisation of ion and small-molecule translocation across lipid bilayers and biological membranes, including those of cells, tissues and vesicles. Solid-state NMR in particular can investigate the interactions and effects of peptides, proteins and small molecules on the structure, phase behaviour and dynamics of lipid bilayers and biological membranes. Similarly, solid-state NMR can elucidate atomic-resolution structure, orientation and dynamics of transmembrane peptides and proteins in lipid bilayers and native biological membranes. Furthermore, the basic NMR experiment is the principle behind magnetic resonance imaging (MRI), which is one of the main established clinical techniques for in vivo imaging of the whole human body and specific organs and tissues. Only certain naturally-occurring nuclei have intrinsic properties that allow them to be used in NMR (and MRI) applications with biological and biomedical systems and samples. NMR active nuclei are those possessing a property called ‘spin’, whereby a charged nucleus spins about an axis and generates its own magnetic dipole moment. This property enables alignment of nuclei in an external magnetic field and absorption of radiofrequency radiation, which is the basis of the NMR experiment (Figure 1).
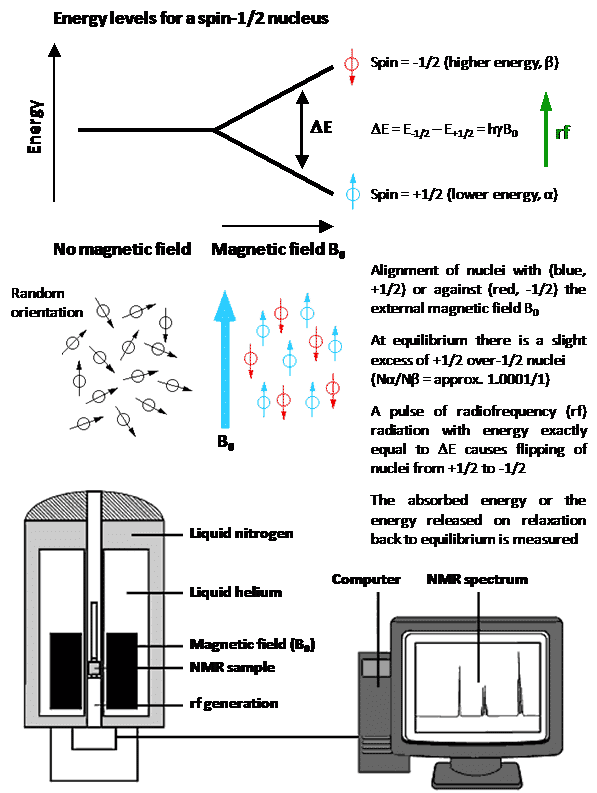
Figure 1. The basic NMR experiment with a spin-1/2 nucleus.
Nuclei that possess an even number of both neutrons and protons have no spin (spin quantum number = 0) and are not NMR-active, for example 2He, 12C, 16O, 32S. Nuclei for which the number of neutrons plus the number of protons is an odd number have a half-integer spin (i.e. spin quantum number = 1/2, 3/2, 5/2, 7/2, 9/2), for example 1H, 13C, 15N, 19F. Nuclei for which the number of neutrons and the number of protons are both odd have an integer spin (i.e. spin quantum number = 1, 2, 3), for example 2H, 6Li, 10B, 14N. In all nuclei for which the spin quantum number is greater than 1/2, the charge distribution of protons is asymmetric (Figure 2), which gives them an electric quadrupole moment in addition to their magnetic dipole moment.
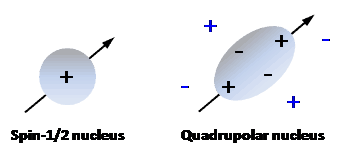
Figure 2. Charge distributions in a spin-1/2 nucleus and in a quadrupolar nucleus.
A spin-1/2 nucleus has a spherical distribution of electric charge. A quadrupolar nucleus has an asymmetric distribution of nucleons, producing a non-spherical positive charge distribution. The nuclear charge distribution (black charges) interacts asymmetrically with electric field gradients (blue charges) in a molecule.
These ‘quadrupolar nuclei’, which constitute over two-thirds of all naturally occurring NMR active nuclei, can have very short longitudinal relaxation times (T1) and produce broad NMR signals or none at all. Quadrupolar nuclei with an integer spin tend to produce much broader signals than those with a half-integer spin. Hence, the most useful nuclei for NMR applications are those with a half-integer spin, especially those with a spin number of 1/2. For a more comprehensive description of nuclear spin systems, the reader is referred to reference [1]. It is fortunate that some of the most common elements found in living organisms have an isotope that is spin-1/2 (i.e. 1H, 13C, 15N, 31P) and these nuclei have prolific use in NMR applications with biological and biomedical systems and samples. The natural background of such nuclei can prove to be a problem for certain NMR studies, however. A surprisingly large number of other nuclei have also been used in published NMR (and MRI) applications with biological and biomedical systems and samples. Table 1 and Figure 3 give properties for 39 such nuclei from 33 different elements that will be covered in this article. Whilst some studies use natural abundance levels of the nucleus being analysed, others require enrichment with the nucleus (isotope labelling) to improve the sensitivity of detection. In the following sections of this article, each of the 39 nuclei is considered in order of increasing atomic number, with details and illustrated examples of published studies, as appropriate.
Element Nucleus Spin number (I)
Natural abundance (%) Chemical shift range (ppm) Sensitivity relative to 1H (enriched) Larmor frequency at 7.05 T (MHz) Larmor frequency at 9.40 T (MHz) Larmor frequency at 11.75 T (MHz)
Hydrogen 1H 1/2 99.99 13 1.00000 300.13 400.13 500.13
2H 1 0.01 13 0.00965 46.07 61.42 76.77
3H 1/2 0.01 13 1.21000 320.13 426.80 533.46
Helium 3He 1/2 0.01 58 0.00348 228.64 304.82 380.99
Lithium 7Li 3/2 92.41 27 0.29400 116.64 155.51 194.37
Boron 10B 3 19.90 110 0.01990 32.25 42.99 53.73
11B 3/2 80.10 110 0.16500 96.29 128.38 160.46
Carbon 13C 1/2 1.11 200 0.01590 75.47 100.61 125.75
Nitrogen 15N 1/2 0.36 900 0.00104 30.42 40.56 50.70
Oxygen 17O 5/2 0.04 1160 0.02910 40.69 54.24 67.80
Fluorine 19F 1/2 100.00 700 0.08320 282.40 376.50 470.59
Sodium 23Na 3/2 100.00 72 0.09270 79.39 105.84 132.29
Magnesium 25Mg 5/2 10.00 70 0.00268 18.37 24.49 30.62
Aluminium 27Al 5/2 100.00 400 0.20700 78.20 104.26 130.32
Silicon 29Si 1/2 4.69 540 0.00786 59.63 79.50 99.36
Phosphorus 31P 1/2 100.00 430 0.06650 121.50 161.98 202.46
Sulphur 33S 3/2 0.75 964 0.00227 23.04 30.71 38.39
Chlorine 35Cl 3/2 75.76 1100 0.00472 29.41 39.20 49.00
37Cl 3/2 24.24 1100 0.00272 24.48 32.63 40.79
Potassium 39K 3/2 93.26 65 0.00051 14.01 18.67 23.34
Calcium 43Ca
7/2 0.14 70 0.00643 20.20 26.93 33.66
Vanadium 51V 7/2 99.75 1900 0.38400 78.94 105.25 131.55
Cobalt 59Co 7/2 100.00 18000 0.27800 71.21 94.94 118.67
Copper 65Cu 3/2 30.85 1100 0.11500 85.25 113.65 142.06
Zinc 67Zn 5/2 4.10 2700 0.00287 18.78 25.04 31.29
Selenium 77Se 1/2 7.63 3000 0.00703 57.24 76.31 95.38
Bromine 79Br 3/2 50.69 600 0.07940 75.20 100.25 125.30
81Br 3/2 49.31 600 0.09950 81.06 108.06 135.07
Krypton 83Kr
9/2 11.50 220 0.00190 11.55 15.40 19.24
Rubidium 87Rb 3/2 27.83 110 0.17700 98.20 130.92 163.65
Cadmium 111Cd
1/2 12.80 650 0.00966 63.64 84.89 106.11
113Cd 1/2 12.22 650 0.01110 66.61 88.80 111.00
Iodine 127I 5/2 100.00 4200 0.09540 60.05 80.06 100.06
Xenon 129Xe 1/2 26.40 500 0.02160 83.47 111.28 139.09
Caesium 133Cs 7/2 100.00 160 0.04840 39.37 52.48 65.60
Platinum 195Pt
1/2 33.83 6700 0.01040 64.52 86.02 107.51
Mercury 199Hg 1/2 16.87 3500 0.00594 53.76 71.67 89.58
Thallium 205Tl 1/2 70.48 7000 0.20200 173.13 230.81 288.49
Lead 207Pb 1/2 22.10 11500 0.00906 62.79 83.71 104.63
Table 1. Properties of NMR active nuclei for biological and biomedical applications.
Natural abundances and Larmor frequencies are given to two decimal places. Sensitivities are given to five decimal places and include the first three non-zero numbers. Information was taken from Bruker NMR frequency tables, the Bruker NMR guide and from the NMR lab website of the Hebrew University of Jerusalem.
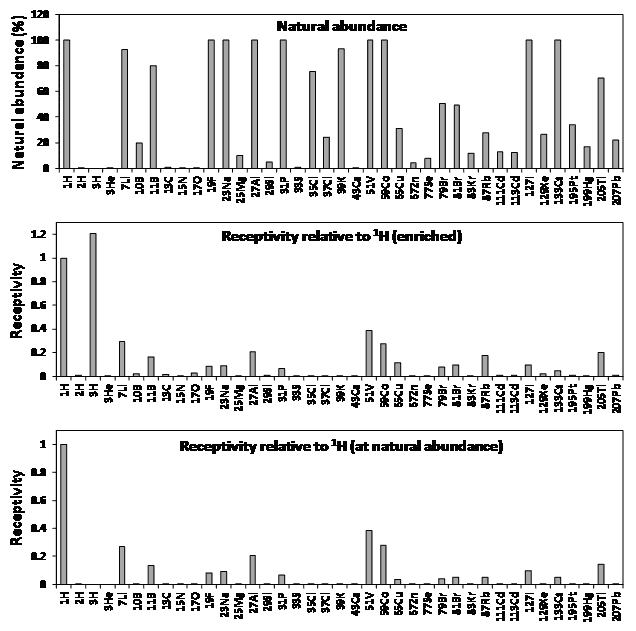
Figure 3. Properties of NMR active nuclei for biological and biomedical applications.
Natural abundances and receptivity values (sensitivity) were taken from Table 1 or calculated using values in Table 1.
2. NMR active nuclei
2.1. Hydrogen (1H, 2H, 3H)
There are three NMR-active isotopes of hydrogen, the spin-1/2 protium (1H), spin-1 deuterium (2H) and spin-1/2 tritium (3H). Whilst 3H is the most sensitive of all NMR active nuclei, it is radioactive (β-emitter), has a very low natural abundance (3 x 10-16%) and is difficult and expensive to obtain or produce. 1H is the most sensitive of all NMR active nuclei after 3H and has a natural abundance of 99.99%, it the most commonly used nucleus for NMR applications and is the nucleus to which all others are compared (Table 1). For example, the receptivity values of 2H, 3H and 13C relative to 1H when enriched are 9.65 x 10-3, 1.21 and 1.59 x 10-2, respectively. Even though the chemical shift range for 1H is relatively small (-1 to 12 ppm), it can produce very sharp and highly resolved signals, depending on sample properties and the NMR pulse sequence used.
2.1.1. Applications of protium (1H)
Whilst 1H is intrinsically involved in a large majority of NMR experiments for biological and biomedical applications, for example in those for structure determination of biomolecules, the large background of hydrogen in these samples means that direct 1H detection has generally not been used. Exceptions include cases where samples have been prepared with various levels of deuteration in order to reduce or eliminate signals originating from background 1H. In addition to removal of interfering 1H signals from NMR spectra, deuteration also eliminates potential proton relaxation pathways and strong dipole-dipole interactions that would otherwise contribute to line broadening effects on the spectra. This is especially important when performing TROSY-type solution-state NMR experiments on detergent-solubilised membrane proteins where deuteration of both the protein and detergent may be essential [2–4]. An increasing number of studies have shown how sample deuteration and/or fast magic angle spinning (MAS) enable high-resolution proton-detected solid-state NMR spectra to be obtained for samples of biological and biomedical origin. Partial deuteration can reduce spectral congestion in 1H, 13C, 15N solid-state NMR correlation spectra [5], thus making structural analysis amenable to larger and more complex biomolecules. Perdeuteration, however, can produce proton-detected solid-state NMR spectra of biomolecules with ultra-high resolution, even at low to moderate (5-30 kHz) MAS frequencies, as demonstrated with samples of amyloid fibrils and membrane proteins [6–8], for example, using the α-spectrin SH3 domain (Figure 4). Using highly deuterated samples, solid-state NMR methods have been developed for sensitivity enhancement by preserving water magnetisation [9] and for resonance assignment using dipolar-based interspin magnetisation transfers [10] and proton-detected 4D experiments [11]. Recent developments in MAS NMR technology have made it possible to spin solid samples up to a frequency of around 110 kHz [12], which improves significantly the feasibility for performing proton-detected measurements. Studies using fast MAS and proton detection have demonstrated resonance assignment procedures and assessments of sensitivity with different protein samples [13–16], quantified sugars in plant tissue [17], investigated structure and dynamics in measles virus nucleocapsids [18] and investigated the organic matrix and monitored structural and dynamic changes in bone [19,20]. Leading-edge technological advances for proton-detected solid-state NMR include fast MAS at ultra-high magnetic field [21] and high-resolution triple resonance micro-MAS NMR with nanolitre sample volumes [22]. All of these ongoing technological developments will make a larger range and complexity of samples of biological and biomedical origin amenable to chemical, structural and dynamic investigation by proton-detected NMR.
Figure 4. High-resolution proton-detected solid-state NMR spectrum of a perdeuterated biomolecule.
A. 1H-detected 1H,15N-correlation spectrum recorded with a perdeuterated α-spectrin SH3 sample that was recrystallised from a buffer containing 90% D2O. B. Amide proton linewidths as a function of MAS rotation frequencies (8-24 kHz) for selected residues. This Figure was reproduced with permission from Reif 2012 [7]; copyright © 2012 by Elsevier Inc.
1H-detected NMR is one of the main methods used for the study of metabolomics (and metabonomics), whereby metabolites in cells, biofluids, tissues or organisms are identified, quantified and changes monitored to reflect the underlying biochemical activity [23]. These can be affected by disease, drugs or environmental variation. The most common human body fluids to be analysed are blood (whole, plasma, serum) and urine [24,25], but also other fluids including cerebrospinal fluid, milk, prostatic fluids, saliva and sweat. Recent studies include 1H and 1H-13C HSQC NMR screening of urine in autism spectrum disorders [26], 1H NMR metabolic profiling of five different brain regions in a mouse model of Alzheimer’s disease [27], 1H NMR assessment of the lipoprotein profile in type 1 diabetes [28], 1H NMR characterisation of serum metabolites in cervical cancer [29], use of 1H NMR to identify urinary biomarkers of severe sepsis and septic shock in the Intensive Care Unit [30] and 1H NMR analysis of metabolic profiles in ovarian tumour cyst fluid [31]. 1H NMR-detected metabolomics is also emerging as a useful tool for assessment of cardiovascular disorders, including cardiac arrest [32,33]. Other types of samples that have been analysed for 1H NMR-detected metabolomics are very diverse and include studies on metabolic profiling of reef-building corals [34] and the brains of sheep exposed to scrapie [35], effects of temperature and diet composition on the early developmental stages of cod larvae [36], predicting the optimum pH for the quality of chicken meat [37] and the detoxification mechanism of cucumber plants exposed to copper nanoparticles [38]. These are just a small number of recent examples, highlighting that a more comprehensive overview of 1H NMR-detected metabolomics applications is beyond the scope of this current work. 1H NMR is also the basis of the well-established and widespread clinical diagnostic tool magnetic resonance imaging (MRI), used for the non-invasive and non-destructive imaging of soft tissues such as brain, heart and muscles and for identifying and monitoring tumors in many organs. Related to both MRI and metabolomics is also the in vivo clinical tool proton magnetic resonance spectroscopy (1H-MRS), which combines 1H NMR-derived metabolic profiles with MRI images to diagnose and monitor a wide range of diseases and conditions.
2.1.2. Applications of deuterium (2H)
The quadrupolar properties of the deuterium nucleus (2H) can produce broad NMR signals of up to a few kHz, resulting in poor resolution. Direct 2H detection is therefore not routinely used for solution-state NMR, but it has found some useful solid-state NMR applications. The relatively low natural abundance of 2H (0.015%) means that 2H-enrichment of samples is usually required. One application is in investigating the structure and phase behaviour of biological membranes [39,40] and their interactions with drugs and antimicrobial peptides using lineshape analysis and relaxation measurements on static samples. This is made possible by the orientation dependence of the 2H electric quadrupolar interaction, which permits the study of molecular orientational order. For example, 2H solid-state NMR has recently been used to investigate the compositional distributions and lipid order profiles of raft model membranes comprising mixtures of site-specifically deuterated N-stearoylsphingomyelins, 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) and cholesterol [41]. The application of 2H solid-state NMR for investigating deformation of lipid bilayers at the atomistic level in liquid-crystalline membranes has been reviewed recently [42]. The effects of commonly used cannabinergic agonists on the lipid membrane bilayer have been investigated using 2H solid-state NMR and hydrated bilayers of dipalmitoyl-phosphatidylcholine (DPPC) deuterated at the 2′ and 16′ positions of both acyl chains. The cannabinergic compounds lowered the phospholipid membrane phase transition temperature, increased the lipid sn-2 chain order parameter at the membrane interface and decreased the order at the centre of the bilayer. It was concluded that compounds can influence lipid membrane domain formation and this may contribute to their cannabinergic activities through lipid membrane microdomain related mechanisms [43]. 2H solid-state NMR studies on the effects of antimicrobial peptides have been performed in model bacterial membranes containing chain-deuterated 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) lipids [44,45] and with membrane-deuterated whole Escherichia coli cells [46]. 2H solid-state NMR is also useful for studying the orientation and dynamics of the peptides themselves in lipid membranes [47]. A high impact example is observation of distinct orientation and dynamics for the drug amantadine in two different binding sites of the M2 proton channel from influenza A virus in 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) bilayers (Figure 5) [48]. Differential binding of deuterated rimantadine enantiomers to the M2 proton channel has also been demonstrated [49].
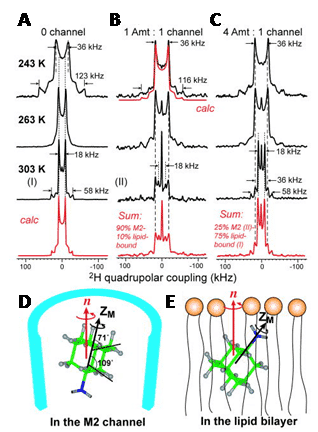
Figure 5. 2H solid-state NMR analysis of amantadine binding to M2 proton channel from influenza A virus.
2H NMR spectra of d15-amantadine in DMPC bilayers as a function of temperature and ratio of amantadine to M2 channel. A. No M2 channel. The calculated spectrum for 303 K reproduces the 1:3 frequency ratio and 4:1 intensity ratio of the two splittings. B. Amantadine/M2 channel ratio = 1:4. The sum spectrum reproduces the 303 K spectrum by 1:9 combination of the lipid-bound 303 K spectrum and peptide-bound 283 K spectrum. C. Amantadine/M2 channel ratio = 4:4. The sum spectrum uses a 1:3 combination of the M2-bound spectrum (II) and lipid-bound spectrum (I). D. Amantadine orientation in the M2 channel. E. One of two possible amantadine orientations in the lipid bilayer. This Figure was reproduced with permission from Cady et al. 2010 [48]; copyright © 2010 by Nature Publishing Group.
Another example is for the cardiac peptide phospholamban, where side-chain and backbone dynamics were measured by lineshape analysis on site-specific deuterated phospholamban in 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers [50]. Dynamics in larger proteins have also been measured using 2H solid-state NMR. For example, in the signalling state of rhodopsin with 11-cis-retinal selectively deuterated at the methyl groups in aligned membranes [51] and in spider dragline silk fibre [52]. Using perdeuterated ubiquitin and Escherichia coli outer membrane protein OmpG as model systems, a suite of three-dimensional 2H-13C correlation experiments for high-resolution solid-state MAS NMR spectroscopy of large proteins were developed by exploiting the favourable lifetime of 2H double-quantum states. The 2H-13C correlation spectra were reminiscent of 1H-13C correlations and allowed a substantial number of assignments for both proteins [53]. A further interesting application of 2H NMR has been in the study of water behaviour in bacterial spores, which can exhibit dormancy and thermal stability under extreme conditions. 2H magnetic relaxation dispersion measurements of water mobility in the core of Bacillus subtilis spores were in support of a gel scenario in which the core is a structured macromolecular framework permeated by mobile water [54]. A separate 2H NMR study suggested that the spore core is more rigid than expected for a gel-like state and that the gel core is inaccessible to external water [55].
2.1.3. Applications of tritium (3H)
Despite having the highest sensitivity of all NMR active nuclei, NMR applications of 3H are scarce, not least because it is radioactive. This is a pity because 3H certainly has some interesting and potentially useful properties. For example, the high gyromagnetic ratio of 3H allows measurement of long-range interatomic distances by solid-state NMR without structural modification of the molecule. Indeed, 3H MAS solid-state NMR has produced the largest NMR distance of 14.4 Å ever measured between two nuclei [56]. Measurement of distances using 3H labels incorporated at specific positions has potential to provide important structural information in samples of biological and biomedical origin. Five 3H NMR studies from the 1990s are described below, but no other significant studies appear to have been reported since then. 3H NMR was used to study anaerobic glycolysis in erythrocytes. Use of [1-3H]-glucose allowed monitoring of the disappearance of α and β tritons and the production of lactate, 1H3HO and some intermediates. Spin-lattice relaxation times (T1) were measured to avoid T1 distortion of the spectral intensities. Formation of 1 mM 1H3HO in the presence of 110 M H2O was detected and this allowed the eventual fate of the label to be observed in vivo [57]. The conformation and dynamics of peptide inhibitor binding to a bacterial collagenase has been studied using 1H and 3H NMR relaxation experiments. Specific 2H and 3H labelling of the succinyl part of the competitive inhibitor succinyl-Pro-Ala allowed measurement of cross-relaxation rates for individual 1H or 3H spin pairs in the inhibitor-collagenase complex and also in the free inhibitor. Determination of order parameters in different parts of the inhibitor indicated that the succinyl and alanyl residues are primarily involved in interactions with the collagenase and that the succinyl moiety adopts a unique trans conformation in the bound state [58]. 3H NMR has been used to study anomeric specificity in complexes of 3H-labelled α- and β-maltodextrins with maltose-binding protein (MBP). At a temperature of 10 ºC, MBP bound α-maltose with 2.7 ± 0.5-fold higher affinity than β-maltose and longer maltodextrins had a ratio of affinities (Kdβ/Kdα) that was significantly greater (10- to 30-fold). Further interpretation of the spectra also revealed how MBP is able to bind both linear and circular maltodextrins [59]. 3H NMR has been used to study two nucleic acid molecules, an 8 kDa DNA oligomer and a 20 kDa ‘hammer-head’ RNA. 3H-1H NOESY experiments allowed observation of through-space interactions in B-form DNA and an unexpected ‘antiphase’ cross-peak at the water frequency. 3H NMR spectra of the RNA molecule indicated conformational dynamics in the conserved region of the molecule in the absence of Mg2+ and spermine, which are two components necessary for cleavage [60]. 3H NMR was used to examine the complex formed by [4-3H]benzenesulphonamide and human carbonic anhydrase I (HCA I), showing that a 1:1 complex exists in solution. Interpretation of 3H relaxation behaviour and 3H-1H NOEs showed that the rate of dissociation of the complex is 0.35 s-1 and that the aromatic ring of the inhibitor undergoes rapid rotation whilst in the complex (Figure 6) [61].
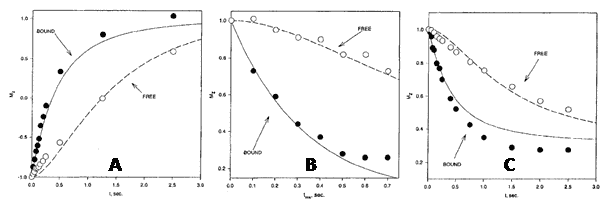
Figure 6. 3H NMR analysis of the complex between [4-3H]benzenesulphonamide and human carbonic anhydrase I.
Comparisons of observed and calculated 3H T1 relaxation behaviour in an inversion-recovery experiment (A), the transient 3H{1H} NOE as a function of mixing time (B) and time development of the 3H{1H} NOE (C) in a sample with HCA I at a concentration of 1.06 mM and a ratio of [4-3H]benzenesulphonamide to HCA I of 1.35:1. This Figure was reproduced with permission from Culf et al. 1997 [61]; copyright © 1997 by Kluwer Academic Publishers.
2.2. Helium (3He)
The spin-1/2 nucleus 3He is very sensitive when enriched, producing sharp signals and has a moderate chemical shift range (-50 to 8 ppm). Other properties of 3He mean the NMR applications of this nucleus are very limited, however. The chemistry of helium is limited to endohedral fullerenes and the resonance frequency of 3He falls outside the range of conventional NMR probes [62], so special equipment is required. 3He also tends to have long relaxation times, with the gas having a T1 of around 1000 seconds. One medical application of 3He NMR that has emerged is the in vivo imaging of lung function. This is made possible because 3He nuclei can be hyperpolarised by spin-exchange optical pumping [63,64]. Thus, the appropriate wavelength of circularly polarised infrared laser light is used to excite electrons in an alkali metal, such as caesium or rubidium, inside a sealed glass vessel. The angular momentum is transferred from the alkali metal electrons to 3He gas nuclei through collisions, which aligns their nuclear spins with the magnetic field to enhance the NMR signal. The resultant hyperpolarised 3He gas can be stored at a pressure of 10 atm for up to 100 hours.
Gas mixtures containing hyperpolarised 3He can be inhaled and then imaged using an MRI scanner to produce pictures of lung and airway function. This method can be used to monitor conditions such as asthma, chronic obstructive pulmonary disease (COPD), cystic fibrosis (CF) and radiation-induced lung injury (Figure 7), and also lung transplantation [65–68].
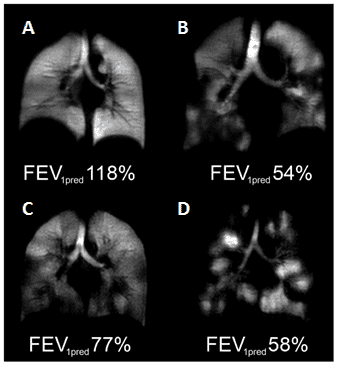
Figure 7. Hyperpolarised 3He-MRI analysis of human lungs.
A. Healthy 45 year old female with predicted forced expiratory volume in the first second of expiration (FEV1pred) 118%. B. COPD 79 year old male with FEV1pred 54%. C. Asthmatic 26 year old male (baseline, no provocation) with FEV1pred 77%. D. CF 23 year old female with FEV1pred 58%. This Figure was reproduced with permission from Fain et al. 2010 [68]; copyright © 2010 by Wiley-Liss, Inc.
2.3. Lithium (7Li)
There are two NMR-active isotopes of lithium, 6Li and 7Li, both of which are quadrupolar. 6Li (natural abundance 7.59%) is spin-1 and produces sharp signals, but has a relatively low quadrupolar moment and low sensitivity. 7Li (natural abundance 92.41%) is spin-3/2, has a higher quadrupolar moment than 6Li and produces broader signals, but it is highly sensitive. The principal use of lithium NMR is 7Li magnetic resonance spectroscopy analysis of the brain for studying and monitoring bipolar disorder, for which lithium and its salts are effective in both acute and prophylactic treatment [69,70]. A 7Li magnetic resonance spectroscopy study of the distribution and regional pharmacokinetics of lithium in rat brain suggested that lithium is most active in a region stretching from the anterior cingulate cortex and striatum to the caudal midbrain, with greatest activity in the preoptic area and hypothalamic region. Some activity was also seen in prefrontal cortex, but only minimal amounts in the cerebellum and metencephalic brainstem [71]. A 7Li and 1H magnetic resonance spectroscopy analysis of the relationship between brain lithium levels and the metabolites N-acetyl aspartate and myo-inositol in the anterior cingulate cortex of older adults with bipolar disorder showed a direct association between brain lithium and higher levels of both metabolites. It was suggested that the higher levels of myo-inositol reflect increased activity of inositol mono-phosphatase [72]. A quantitative 7Li magnetic resonance spectroscopy study of the normal human brain measured the in vivo T1 of 7Li as 2.1 ± 0.7 seconds. The mean brain 7Li concentration was 0.71 ± 0.1 mM, with no significant difference between grey and white matter, and the mean serum concentration was 0.9 ± 0.16 mM [73]. A later quantitative study on bipolar patients stable on long-term lithium treatment demonstrated a bi-exponential lithium T2 relaxation in the majority of cases with an average short decay time of 5.3 ± 1.4 ms and an average long decay time of 68.2 ± 10.2 ms. In two of the patients, a strongly mono-exponential T2 relaxation was observed with an average decay time of 47.4 ± 1.3 ms [74]. The compartmental distribution of lithium as a function of total lithium concentration in rat brain was studied using these biexponential 7Li T2 decays. A linear interpolation using the biexponential T2 values to estimate intracellular lithium from individual monoexponential T2 decays were also assessed. The intracellular T2 was 14.8 ± 4.3 ms and the extracellular T2 was 295 ± 61 ms. The fraction of intracellular brain lithium ranged from 37.3 to 64.8% (mean 54.5 ± 6.7%) and did not correlate with total lithium concentration. The estimated intracellular lithium concentration ranged from 47 to 80% (mean 68.3 ± 8.5%) of the total brain lithium and was highly correlated with it [75]. A quantitative 7Li magnetic resonance spectroscopy study of brain lithium levels after six weeks of lithium therapy in patients with bipolar disorder revealed a significant association between central and peripheral lithium levels in remitters but not in non-remitters. It was therefore suggested that non-remitters may not transport lithium properly to the brain and this may underlie resistance to treatment with lithium. Also, brain lithium (but not plasma lithium) was inversely correlated with age, whilst plasma lithium did not correlate with any clinical outcome, lithium dosage or adverse effects [76]. The putative target protein for lithium therapy in bipolar disorder is inositol monophosphatase, which catalyses the hydrolysis of inositol monophosphate to inorganic phosphate and inositol. Using 7Li MAS solid-state NMR, including 13C-7Li dipolar recoupling experiments (Figure 8), the bound form of lithium in the active site of an inositol monophosphatase from Escherichia coli (SuhB) has been observed [77]. Lithium binds to site II in SuhB that is coupled to three aspartate residues (84, 87, and 212). The inositol monophosphatase activity of SuhB is strongly inhibited by lithium and SuhB shares significant sequence similarity with human inositol monophosphatase, including most of its key active-site residues.
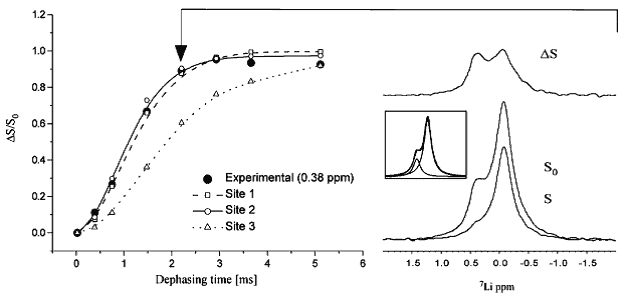
Figure 8. 13C-7Li dipolar recoupling analysis of lithium binding to an Escherichia coli inositol monophosphatase (SuhB). Comparison of experimental CP-filtered, 7Li-detected REDOR data points (ΔS/S0) of the 0.38 ppm 7Li signal from fully labelled SuhB inositol monophosphatase with Spinevolution simulations of the three Mg2+ binding sites from bovine inositol monophosphatase (PDB entry 2BJI). For sites I and II, three carboxyl carbons and their three nearest carbons were used in a seven-spin simulation. Site I: Glu-70 Cδ and Cγ, Asp-90 Cγ and Cβ, and Ile-92 C′ and Cα. Site II: Asp-90/93/220 Cγ and Cβ. For site III, simulations were performed using all four carbons within 5 Å of this site (Lys-36 Cε, Asp-41 Cγ, Glu-70 Cδ and Cγ). Typical 7Li spectra, corresponding to S and S0 at the time point indicated by the arrow, are shown at the right along with the difference spectrum ΔS; the peak deconvolution of S0 is shown inset. This Figure was reproduced with permission from Haimovich et al. 2012 [77]; copyright © 2012 by American Chemical Society.
2.4. Boron (10B, 11B)
Boron has two NMR-active isotopes, spin-3 10B (natural abundance 19.9%) and spin-3/2 11B (natural abundance 80.1%). 11B is the preferred nucleus for NMR applications because it has a lower quadrupole moment and is more sensitive than 10B. A common problem encountered in boron NMR spectroscopy is broad background signals originating from regular NMR tubes (made of borosilicate glass) and from probe components. These can be avoided by using quartz tubes, which are more expensive, and/or by using a DEPTH pulse sequence, which increases the signal-to-background ratio [78]. The most common application of boron NMR is for the detection of 10B and 11B in boron neutron capture therapy (BNCT). In this process of cancer treatment, boron-containing molecules enriched with 10B are targeted to the tumour and irradiated with thermal or epithermal neutrons. Capture of the neutrons by 10B nuclei generates cell-damaging radiation (production of an α-particle and a 7Li-particle) that is confined to single-cell dimensions. Boron NMR is used to study the metabolism and pharmacokinetics of the boron-containing molecules and for non-invasive in vivo mapping of the molecules [79,80]. Preliminary 1H and 10B NMR relaxation studies have been performed in animal tissues and in living tumour cells to assess the suitability of 10B molecules tagged with a Gd(III) paramagnetic ion for BNCT. Such molecules may be useful as contrasting agents in MRI for mapping boron distribution in tissues [81]. 10B NMR has also been used to achieve the simultaneous analysis of 10B-p-boronophenylalanine (BPA), 10B-BPA-fructose complex and total 10B in blood for BNCT studies [82]. In an interesting application, 11B MAS solid-state NMR has been used to assess pH levels in red coralline algae by measuring boron isotopic compositions. In this respect, 30% of boron in powdered bulk samples was present as boric acid [83]. The isotopic composition and elemental abundance of boron in marine carbonates is a useful tool for tracking changes in seawater pH and carbonate chemistry.
2.5. Carbon (13C)
The spin-1/2 nucleus 13C (natural abundance 1.11%) is the only NMR-active isotope of carbon. 13C has relatively low sensitivity and usually requires enrichment, but it produces sharp signals and has a wide chemical shift range (0 to 200 ppm) that allow good spectral dispersion. After 1H, 13C is one of the most commonly used nuclei for biological and biomedical NMR applications, for example, in protein structure determination, observation and quantification of ligand and drug binding, characterisation of protein-ligand and protein-protein interactions and measurement of kinetics and dynamics. Despite the widespread involvement of 13C in NMR applications, direct 13C detection has only recently become useful for studying biomolecules. Developments include spin-state-selective methods that achieve homonuclear decoupling in the direct acquisition dimension of 13C detection and high-resolution methyl-selective 13C NMR experiments in both the solution- and solid-state [84,85]. Methods have also been developed for direct-detection 13C biomolecular NMR spectroscopy in living cells and other in vivo methods [86,87]. Hence, 13C has proved to be a very useful NMR-active nucleus for metabolomics, whereby the metabolic fluxes of 13C-enriched or natural abundance substrates can be monitored in vivo. A principal application of 13C NMR is in analysis of brain metabolism [88,89]. For example, the ratio of acetate-to-glucose oxidation in astrocytes has been measured from a single 13C NMR spectrum of cerebral cortex [90]. Along with 1H magnetic resonance spectroscopy, 13C magnetic resonance spectroscopy is useful for monitoring the glutamate-glutamine cycle in the brain and central nervous system of healthy individuals and of those with neurological disorders in which this cycle is disturbed [91,92].
In addition to [18F]FDG-positron emission tomography (FDG-PET) [93], 13C magnetic resonance spectroscopy is a principal technique used to assess glucose transport and metabolism following traumatic brain injury (Figure 9) [94–96]. In a recent study, 13C NMR was used to explore neuron-astrocyte metabolic cooperation in the brains of diabetic mice with cognitive decline using intravenous infusions of [2-13C]-acetate and [3-13C]-lactate.
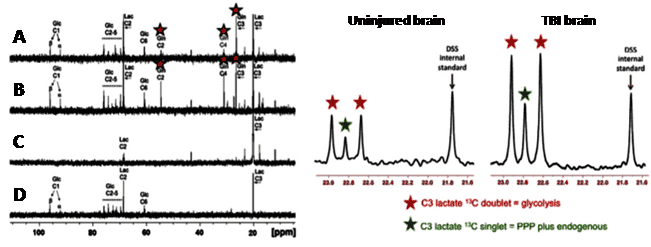
Figure 9. 13C magnetic resonance spectroscopy analysis of glucose transport and metabolism following traumatic brain injury. 13C-NMR spectra achieved by ex vivo NMR analysis of microdialysate after delivery of [3-13C]lactate and [1,2-13C2]glucose to traumatic brain injury (TBI) patients.
Left panel: A and B. Examples of 13C NMR spectra of brain microdialysates from a TBI patient receiving perfusion with [3-13C]lactate (4 mM) by microdialysis catheters via a craniotomy; red stars indicate 13C signals for glutamine C4, C3 and C2 indicating metabolism via the TCA cycle. C. 13C NMR spectrum of the [3-13C] lactate substrate solution prior to perfusing. D. 13C NMR spectrum of brain microdialysate from an unlabelled patient. This Figure was reproduced with permission from Gallagher et al. 2009 [95]; copyright Ó 2009 by Gallagher et al. Right panel: Examples of 13C NMR spectra of brain microdialysates from patients receiving [1,2-13C2]glucose (4 mM) perfused via a microdialysis catheter. The uninjured brain is a normal-appearing brain in a patient operated on for a benign tumour elsewhere in the brain. The TBI brain is from a patient with a diffuse injury. The part of the spectrum illustrated in each case is for the C3 carbon of lactate. Also present in this part of the spectrum is one of the signals due to the internal standard DSS (2,2-dimethyl-2-silapentane-5-sulfonate sodium salt). The remainder of the spectrum (including the main DSS signal at 0 ppm) is not shown. The C3 doublet indicated by red stars represents lactate doubly labelled with 13C, produced by glycolysis; the C3 signal for 13C is split into 2 peaks by coupling to 13C also present at the neighbouring C2 position within the same molecule. The C3 singlet indicated by green stars represents lactate singly labelled with 13C, produced via the pentose-phosphate pathway. This Figure was reproduced with permission from Carpenter et al. 2014 [96]; copyright © 2014 by Carpenter et al.
Relative to wild-type mice, the diabetic mice had significantly lower 13C labelling in neurotransmitters including glutamine, glutamate, and γ-aminobutyric acid after [2-13C]-acetate infusion, whilst infusion of [3-13C]-lactate resulted in increased 13C-enrichment of neurotransmitters in the diabetic mice. The results indicated a possible disturbance of neurotransmitter metabolism during the development of cognitive decline in diabetics [97]. The 13C nucleus plays important roles in a range of solid-state NMR applications. This includes structure determination of membrane proteins, which are major molecular targets for drug discovery. Structural and dynamic information on membrane proteins can be obtained from static oriented samples or from samples subjected to MAS in native lipid membranes [98]. Introduction of 13C labels at specific sites in the proteins allows measurement of highly accurate through-space distances using homonuclear (13C-13C) and heteronuclear (e.g. 13C-15N, 13C-19F) dipolar recoupling experiments such as rotational resonance [99] and rotational echo double resonance (REDOR) [100], respectively, and derivatives thereof. Under favourable sample and dynamic conditions, the 1H-13C cross-polarisation magic-angle spinning (CP-MAS) experiment can be used to observe and quantify the binding of 13C-enriched ligands and drug molecules to wild-type and mutant membrane proteins in native membranes [101–104]. Measurement of through-space inter-atomic distances and torsion angles can then allow elucidation of atomic resolution structures, molecular conformations, positions and orientations of ligands and drugs in their binding sites [104–111]. For example, 13C-2H REDOR NMR was used to define the structure of the amantadine binding site of the M2 proton channel from influenza A virus in lipid bilayers (Figure 10) [48].
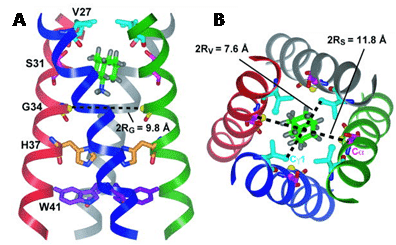
Figure 10. Solid-state NMR structure of amantadine-bound M2 proton channel in lipid bilayers. A. Side view showing Ser31, Val27, Gly34, His37, Trp41 and amantadine in the high-affinity luminal site. Ser31 Cα lies in the mid-plane between the two rings of deuterons. The instantaneous orientation of amantadine, which is slightly tilted from the channel axis, is shown. The time-averaged amantadine orientation is parallel to the channel axis. B. Top view showing the Ser31 and Val27 pore radii. This Figure was reproduced with permission from Cady et al. 2010 [48]; copyright © 2010 by Nature Publishing Group.
Such measurements can contribute important information for the design and discovery of drugs involving membrane-embedded targets [112–116]. 13C solid-state NMR has also provided chemical, structural and dynamic characterisation of a wide range of other types of samples of biological and biomedical origin. These include amyloid fibrils from the amyloid plaques that are a hallmark of Alzheimer’s disease [117–119], structural proteins such as elastin [120] and bone implants [121]. Characterisations of plant material include the structure and dynamics of pectic polysaccharides and the structure of cellulose and its interactions with matrix polysaccharides in plant primary cell walls [122,123], fungal degradation in wood by monitoring lignin and cellulose composition [124] and chemical composition in the biomass of a mushroom [125]. Variable-temperature 13C solid-state NMR has been used to study molecular structures of honey-bee wax and silk [126]. 1H-13C CP-MAS solid-state NMR has also been used to study different types of soil samples and effects of environmental factors on their composition [127–129].
2.6. Nitrogen (15N)
There are two NMR-active isotopes of nitrogen, spin-1 14N (natural abundance 99.63%) and spin-1/2 15N (natural abundance 0.37%). 14N has medium sensitivity, but its quadrupolar properties produce broad signals that are usually unobservable by a high-resolution NMR spectrometer. 15N is very insensitive, but its sharp lines and wide chemical shift range (0 to 900 ppm) can produce well-dispersed spectra. Hence, 15N-enriched samples are widely used for determining the backbone structures of proteins by NMR. This includes the basic solution-state [15N,1H]-HSQC and [15N,1H]-TROSY experiments and the plethora of correlation experiments used for making resonance assignments and structural measurements. Because of the low gyromagnetic ratio of 15N, the direct detection of 15N has rarely been used in multidimensional NMR experiments on proteins. It has recently been shown how selection of the TROSY components of proton-attached 15N nuclei can produce high quality 15N-detected spectra in high field magnets (>600 MHz) by taking advantage of the slow 15N transverse relaxation and compensating for the inherently low 15N sensitivity. 15N-detected TROSY experiments theoretically produce the narrowest linewidths at a magnetic field of 900 MHz and sensitivity reaches a maximum at around 1.2 GHz [130]. Indeed, it was then demonstrated that 15N-detected TROSY produces comparable sensitivity to 1H-detected TROSY for non-deuterated, large proteins under physiological salt conditions (Figure 11) [131]. The 15N nucleus is also used for solid-state NMR structural and dynamics measurements on proteins and other biomolecules in a similar manner to those with 13C.
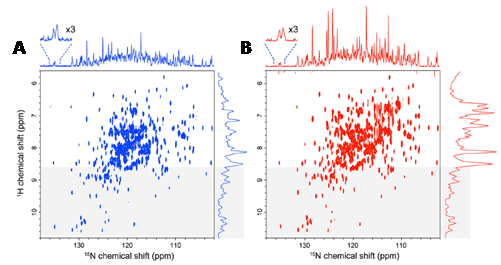
Figure 11. High quality 15N-detected TROSY spectrum of a non-deuterated, large protein under physiological salt conditions. Comparison of a 15N-detected TROSY-HSQC spectrum (A) and a 1H-detected TROSY-HSQC spectrum (B) of 0.5 mM non-deuterated maltose binding protein in complex with 2 mM β-cyclodextrin at 283 K. A. The 15N-detected TROSY- HSQC was recorded in 8.5 h, ns = 176, F1 = 128 pts (14 ms), F2 = 2048 pts (315 ms). B. The 1H-detected TROSY-HSQC was recorded in 8.5 h, ns = 12, F1 = 2048 pts (315 ms), F2 = 800 pts (18 ms). The 1H-detected TROSY-HSQC was transposed. 15N and 1H projections of the 2D spectra are indicated without any multiplication. The regions that were expected to contain mainly the resonances from the structured region of the proteins (8.6 ppm in the 1H dimension) are indicated by grey shading. The spectra were recorded at 283 K and 800 MHz, and the apparent sc of the system deduced from the TROSY for rotational correlation times (TRACT) experiment was 35 ns. This Figure was reproduced with permission from Takeuchi et al. 2016 [131]; copyright ©2016 by Springer Science+Business Media Dordrecht.
2.7. Oxygen (17O)
The only NMR active isotope of oxygen is the spin-5/2 17O (natural abundance 0.038%). 17O-enrichment is generally required for NMR studies, which is very expensive, and the broad signals produced by 17O are partly compensated for by its wide chemical shift range (-40 to 1120 ppm). The quadrupolar moment of 17O can interact with local electric field gradients, resulting in extremely short T1 and T2 relaxation times in the order of several milliseconds. Solution-state NMR applications of 17O with biomolecules are very limited, one exception is a study that used 17O NMR for observing an oxidised cysteine residue in Cu,Zn-superoxide dismutase [132]. Solution-state 17O NMR has found a useful application in the study of brain function and cerebral bioenergetics, however. Cerebral blood flow can be studied by monitoring washout of the tracer H217O in brain tissue following an intravascular bolus injection of 17O-labelled water. The cerebral metabolic rate of oxygen utilisation can be measured by monitoring the dynamic changes of metabolically generated H217O in brain tissue from inhaled 17O-labelled oxygen gas [133,134]. Similarly, 17O-MRI has been developed for monitoring oxygen consumption in the heart, brain and in tumours by detecting metabolically generated H217O following injection of a suitable 17O-labelled tracer into blood or inhalation of 17O-labelled oxygen gas [135–137]. Experiments have also demonstrated that direct cerebral and cardiac 17O-MRI at 3 Tesla are feasible using natural abundance 17O. In the brain, a signal-to-noise ratio of 36 was obtained at a nominal resolution of 5.6 mm3 and with a T2 relaxation time of 1.9 ± 0.2 ms; in the heart, 17O images were acquired with a temporal resolution of 200 ms [138]. A few studies have shown that it is feasible to apply high-field 17O solid-state NMR to biomolecules, including membrane-embedded peptides and proteins. The first example used a selectively 17O-labelled transmembrane peptide, 17O-[Ala12]-WALP23, in hydrated phospholipid vesicles, from which distance restraints were estimated [139]. A complete resolution of the eight non-equivalent oxygen sites in monosodium L-glutamate monohydrate was demonstrated by 17O NMR with 1H-decoupled double angle rotation and multiple quantum experiments [140,141]. 17O MAS analysis of the tetrameric 28-residue phospholemman transmembrane domain, in which one glycine residue (Gly14) from each helix was enriched to <40% 17O, suggested that the tetramer is an asymmetric unit with either C2- or C1-rotational symmetry along the bilayer normal [142]. Hydrogen bonding in Alzheimer’s amyloid-β fibrils has been probed by 15N-17O REAPDOR solid-state NMR [143]. These studies have shown that sufficient resolution and sensitivity can be achieved in 17O solid-state NMR experiments with samples that contain either 100% or dilute 17O enrichment, therefore showing potential for wider biological and biomedical applications.
2.8. Fluorine (19F)
The spin-1/2 nucleus 19F (natural abundance 100%) is very sensitive, produces sharp signals and has a wide chemical shift range (-300 to 400 ppm). Indeed, 19F is of similar size and behaves similarly to 1H and the receptivity of 19F relative to 1H is x0.83, making 19F the third most sensitive of all NMR active nuclei after 3H and 1H. 19F is therefore a useful NMR nucleus for biological and biomedical applications, where samples usually contain a very low background of fluorine or none at all. 19F solution-state NMR can be used to study interactions of peptides and proteins, protein conformation and dynamics, ligand and drug binding and for drug discovery. Such studies have been assisted by advances in biosynthetic methods and fluorine chemistry for introducing fluorine into peptides, proteins and small molecules with high precision, and by technological advances in NMR spectrometers and probe design for increasing the sensitivity of 19F detection [144–146]. For example, 19F solution-state NMR has been used to study conformational change in the dengue virus NS2B-NS3 protease, which is an antiviral target for drug development. 19F NMR measurements showed that low pH or the binding of bovine pancreatic trypsin inhibitor promote a conformation change from an open to a closed state, thus demonstrating an importance of charge forces in the interaction between NS2Bc and NS3p. A mutation (H51A) impaired the charge interaction and the pH dependence of the conformational changes and stabilised the open conformation, whilst the addition of the inhibitor still converted NS2B-NS3p from an open to a closed state [147]. The introduction of fluorine-containing amino acids into proteins allows 19F-observed NMR to be used in small molecule drug discovery, whereby the binding of molecules is detected by changes in the 19F signals [148]. For example, a fragment screening and druggability assessment of the CREB binding protein/p300 KIX domain was performed using protein-observed 19F NMR spectroscopy. Fluorination of aromatic side chains allowed the screening of 508 compounds and validation by 1H-15N HSQC NMR spectroscopy led to identification of a minimal pharmacaphore for the MLL-KIX interaction site [149]. 19F NMR drug screening strategies do not always require isolation of the target protein, but can also be performed on intact living cells and cell extracts. For example, a 19F NMR-based assay called ‘n-FABS’ (n-fluorine atoms for biochemical screening) in living mammalian cells expressing the membrane protein fatty acid amide hydrolase has been developed. The method allows identification of both weak and potent inhibitors and measurement of their potency in a physiological environment [150]. The almost absence of 19F in the body makes 19F-MRI a useful tool where appropriate 19F tracers can be introduced. High-field 19F-MRI has been shown as potentially useful for the imaging of amyloid-β in senile plaques during the pathological development of Alzheimer’s disease. Three 19F tracers allowed detection of amyloid deposition in the brain of transgenic mouse models of Alzheimer’s disease; these are (E,E)-1-fluoro-2,5-bis-(3-hydroxycarbonyl-4-hydroxy)styrylbenzene (FSB), 1,7-bis(4′-hydroxy-3′-trifluoromethoxyphenyl)-4-methoxy-carbonylethyl-1,6-heptadiene3,5-dione (FMeC1, Shiga-Y5) and 6-(3′,6′,9′,15′,18′,21′-heptaoxa-23′,23′,23′-trifluoro-tricosanyloxy)-2-(4′-dimethylaminostyryl)-benzoxazole (XP7, Shiga-X22) [151]. 19F-MRI can be used for in vivo cell tracking by using suitable 19F-compounds together with specifically adapted hardware and acquisition methods [152]. For example, the fate of 19F-labelled mesenchymal stem cells have been tracked in a mouse model [153] and 19F-labelled stromal vascular fraction cells have been tracked as part of a human phase I trial during treatment of radiation-induced fibrosis in breast cancer patients [154]. 19F-MRI can also be used for tracking the biodistribution of 19F-labelled drugs [155] and for real-time gastrointestinal tracking of 19F-labelled catheter devices [156]. 19F solid-state NMR has largely been used to measure distances, conformations and dynamics in site-specific 19F-labelled membrane peptides and proteins. The high gyromagnetic ratio of the 19F nucleus makes it suitable for measuring relatively long internuclear distances using dipolar recoupling experiments such as REDOR. For example, a modified REDOR sequence involving 1H homonuclear decoupling and composite 19F pulses can measure 1H-19F distances to approximately 8 Å and this was used to measure a [1HN]Leu-[19F]Phe peptide distance of 7.7 Å [157]. 19F spin diffusion solid-state NMR experiments measured a distance of approximately 11 Å between nearest neighbour [19F]Trp41 residues in the tetrameric helical bundle of the M2 proton channel from influenza A virus, confirming its side-chain conformation [158]. By monitoring pH-dependent differences in 19F dipolar couplings and motionally narrowed chemical shift anisotropies of the [6-19F]Trp41 residue, further 19F solid-state NMR measurements demonstrated how Trp41 participates in the gating mechanism of the same channel (Figure 12) [159].
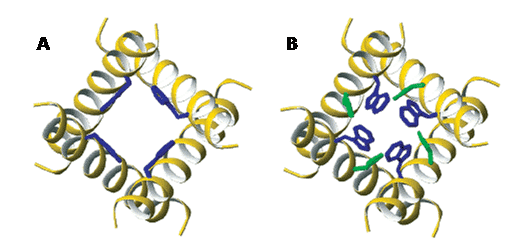
Figure 12. Side chain conformations of Trp41 and His37 in the transmembrane domain of the M2 proton channel from influenza A virus. A. The activated state at pH 5.3 for Trp41 (blue) (ø1, ø2) (-50°, +115°). His37 torsion angles are not defined. B. New coordinates for the inactivated state at pH 8.0 for His37 (green) (-175°, -170°) and Trp41 (-100°, +110°) based on 19F solid-state NMR measurements. This Figure was reproduced with permission from Witter et al. 2008 [159]; copyright © 2008 by American Chemical Society.
Using conformationally constrained 19F-labelled amino acids, it is possible to double the accessible 19F-19F internuclear distance range by combining a highly sensitive solid-state multipulse 19F-NMR scheme with favourable sample geometry. Two rigid 4F-phenylglycine labels placed into a helical antimicrobial peptide embedded in fluid oriented membrane samples and a modified Carr-Purcell-Meiboom-Gill sequence produced an intramolecular distance of 6.6 Å for the labels spanning one helix turn, and 11.0 Å when the labels spanned two turns [160]. 13C/15N-19F REDOR NMR has been used to study the interaction of HIV TAR RNA with the viral regulatory peptide Tat. A critical arginine in the peptide was uniformly 13C and 15N labelled and 5-fluorouridine was incorporated at the U23 position of the TAR RNA. [5-19F]U23-13C and [5-19F]U23-15N distances were in good agreement with distances obtained from solution-state NMR structures of partial complexes of Tat with TAR RNA [161]. 19F MAS solid-state NMR experiments have been performed on diacylglycerol kinase (DAGK) site-specific labelled with trifluoromethyl-phenylalanine in native lipid membranes. In comparison with solution-state NMR data of purified DAGK in detergent micelles, the MAS NMR data showed how 19F chemical shifts of residues at different membrane protein locations were influenced by interactions between membrane proteins and their surrounding lipid environments. Meanwhile, 19F side chain longitudinal relaxation values were likely affected by different interactions of DAGK with the planar lipid bilayer compared with globular detergent micelles [162]. In other types of samples, the molecular conformation, membrane alignment and dynamic behaviour of the cationic peptide [KIGAKI]3, which serves as a model for amyloid-like β-sheet aggregation in membranes, were characterised by 19F solid-state NMR in DMPC lipid bilayers. The data demonstrated a concentration-dependent transition from monomeric β-strands to oligomeric β-sheet amyloid-like fibrils [163]. The orientation of 6-F-cholesterol in DMPC lipid bilayers was analysed by combined use of 19F chemical shift anisotropy, 2H NMR, and 13C-19F REDOR experiments. The data suggested that the conformational and dynamic properties of 6-F-cholesterol in DMPC lipid bilayers are similar to those of unmodified cholesterol [164].
2.9. Sodium (23Na)
The only naturally occurring isotope of sodium is the spin-3/2 23Na, which is the second most abundant NMR-active nucleus in living tissues after 1H. The two main uses of 23Na in biological and biomedical NMR applications are measurement of sodium translocation across membranes and 23Na-MRI. 23Na NMR can be used to measure the translocation of sodium ions across membranes in cells, organelles and liposomes using a membrane-impermeable chemical shift reagent to resolve the signals originating from internal and external sodium. Commonly used shift reagents are dysprosium (III) tripolyphosphate [Dy(PPP)2(7-)] and thulium 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetrakis-(methylene phosphonate) [TmDOTP] [165,166]. In erythrocytes, measurement of sodium translocation by 23Na NMR has been used as an assay of Na+/K+-ATPase activity [167], for measuring the maximal rate of active sodium ion efflux at 10.1 ± 1.0 mM/hour/litre of cells [168] and for studying Na+-K+ co-transport [169]. A 23Na NMR study of the intracellular sodium concentration in the amoebae from the slime mold Dictyostelium discoideum showed that it remained low (0.6 to 3.0 mM) in the presence of external sodium (20 to 70 mM) and a large sodium gradient (20- to 40-fold) was maintained. Introduction of nystatin, an antibiotic known to perturb the ion permeability of membranes, increased the intracellular sodium concentration [170]. 23Na NMR was to measure the intracellular sodium concentration and sodium transport in Escherichia coli cells under both aerobic and glycolytic conditions. Sodium efflux and maintenance of a stable low intracellular sodium concentration correlated with development and maintenance of the proton motive force, which is consistent with proton-driven Na+/H+ exchange as the mechanism of sodium transport [171]. 23Na NMR measurements of sodium transport in cells of Bacillus subtilis defect in the Mrp (multiple resistance and pH) antiporter complex correlated to the inability of this strain to maintain a lower internal sodium concentration than an external one [172]. 23Na-MRI can be used for imaging of sodium in the brain (Figure 13) and in the heart. See references [173, 174] for recent reviews.
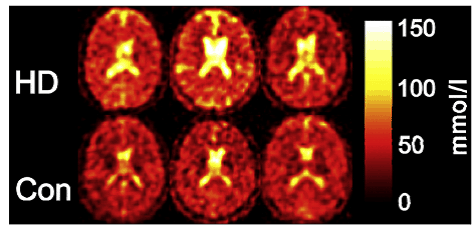
Figure 13. 23Na-MRI analysis of human brain. The total sodium content of the brain of subjects suffering from Huntington’s disease (HD) is increased compared with healthy controls (Con) of the same age and sex. This Figure was reproduced with permission from Reetz et al. 2012 [175]; copyright © 2012 by Elsevier Inc.
A lower sensitivity and significantly reduced multi-component relaxation behaviour (T1 ~30 ms, T2 ~0.5-4 ms and 12-20 ms) of 23Na compared with 1H mean that use of sodium nuclei for imaging is more challenging. Hence, in vivo sodium imaging is feasible only at high magnetic field strength and by use of fast, specialised and ultra-short echo-time MRI pulse sequences. Also, intra- and extra-cellular sodium cannot be unambiguously resolved without the use of potentially toxic chemical shift reagents. 23Na-MRI has demonstrated a correlation between sodium accumulation in brain and disability in relapsing-remitting multiple sclerosis. The study involved acquisition of three-dimensional imaging data at 3.0 T to quantify total sodium concentration levels within specific compartments (multiple sclerosis lesions, white matter and grey matter). Statistical mapping analysis showed confined increases in total sodium concentration inside the brainstem, cerebellum and temporal poles in early relapsing-remitting multiple sclerosis and widespread increases affecting the entire brain in advanced relapsing-remitting multiple sclerosis [176]. 23Na-MRI has also demonstrated heterogeneity of total sodium concentration in hyperacute, acute and chronic lesions in multiple sclerosis [177]. 23Na-MRI in vivo analysis at 3.0 T of human lumbar vertebral discs in healthy volunteers and in patients with low back pain showed decreases in intervertabral disc sodium content with disc degeneration [178]. A 23Na-MRI study of the lower leg in acute heart failure patients during diuretic treatment showed that plasma sodium levels did not change during therapy, whilst sodium concentrations in muscle and skin decreased after furosemide therapy. Sodium concentrations in muscle and skin of patients before and after diuretic therapy were significantly higher than in healthy subjects [179].
2.10. Magnesium (25Mg)
The only naturally occurring NMR-active isotope of magnesium is the spin-5/2 25Mg (natural abundance 10.0%), which has low sensitivity and produces moderately broad signals. The biological and biomedical applications of 25Mg NMR are relatively scarce. 25Mg NMR has been used to study magnesium binding to the erythrocyte constituents ATP, ADP, AMP, 2,3-bisphosphyoglycerate (DPG), and hemoglobin, thus demonstrating a possible method for studying the binding of magnesium within erythrocytes and other cells [180]. 25Mg NMR was used to study interactions of the activating cations with their respective binding sites in the enzymes yeast enolase and rabbit muscle pyruvate kinase [181]. 25Mg NMR was also used to study the divalent metal binding sites of of NAD+-dependent and NADP+-dependent isocitrate dehydrogenases from pig heart. Data showed that magnesium has a similar dissociation constant (1.8 mM) from NADP+-dependent isocitrate dehydrogenase as from the enzyme-isocitrate complex (1.1 mM). The extrapolated linewidth of bound magnesium increased from 674 Hz in the binary complex to 10,200 Hz in the ternary complex. Hence, the quadrupole coupling constant, calculated from relaxation rates, is larger in the ternary complex, which indicated greater distortion in the magnesium coordination sphere. Results were consistent with the metal sites having anisotropic octahedral symmetry [182]. 25Mg NMR was used to study the polyelectrolyte behaviour of actin filaments. Data from F-actin solutions showed that the rotational correlation times of magnesium ions are independent of the overall rotational dynamics of the actin filaments, and competitive binding experiments demonstrated a facile displacement of F-actin-bound magnesium by [Co(NH3)6]3+. ATP also competed effectively with F-actin filaments for binding to magnesium ions. The results supported the hypothesis that magnesium ions bind loosely and non-specifically to F-actin filaments and therefore show behaviour typical of counterions in polyelectrolyte solutions [183]. 25Mg NMR has contributed to a study that revealed the stacking of bacteriochlorophyll c macrocycles in chlorosome from Chlorobium limicola. The dimer-based stacking of the macrocycles was demonstrated by 25Mg NMR, which exhibited a pair of signals showing different quadrupole coupling due to the presence or absence of a water molecule in the axial position [184]. 25Mg solid-state NMR has been used to characterise magnesium binding to the DNA repair protein apurinic/apyrimidic endonuclease 1 (APE1). Low-temperature measurements and various mutants of APE1 were used to demonstrate that magnesium ions bind to APE1 and a functional APE1-substrate DNA complex with an overall stoichiometry of one magnesium ion per mole of APE1. The NMR spectra also showed that the single magnesium ion site is disordered and this is likely due to the arrangement of the protein-ligands (cis and trans isomers) about the magnesium ion [185].
2.11. Aluminium (27Al)
The spin-5/2 nucleus 27Al (natural abundance 100%) has relatively high sensitivity but produces broad signals. Biological and biomedical applications of 27Al NMR are relatively rare and varied. The earliest of these used 27Al NMR to investigate the binding of aluminium to [Leu5]-enkephalin. The spectra suggested that aluminium ions binds at two metal-binding sites, the first of which involves the Tyr1CO and Leu5COO– groups to give a 2:1 species in a tetracoordinated structure. Binding of aluminium at the second site involves the NH2 terminal groups of the tyrosine moiety in a 2:2 species and 27Al chemical shift values strongly suggested that this aluminium atom displays an octahedral environment [186]. A later study demonstrated the potential of 27Al NMR for in vivo analysis and imaging of the human gastric lumen. This was based on following the dissolution kinetics of aluminium-containing drugs at physiological doses and their removal from the human stomach. Aluminium concentrations as low as 0.5 mg could be detected and the time course of gastric emptying was visualised with 27Al-MRI under normal conditions and in the presence of an antimuscarinic agent, which reduces gastric motor function [187]. Gastric emptying and gastrointestinal transit times in mice and humans have also been monitored by 27Al NMR. This study used orally administered aluminium bound to ion-exchange resin and perfluorononane as selective and specific markers for the stomach and the entire gastrointestinal tract, respectively [188]. 27Al NMR has been used to study the transport of aluminium across yeast cell membranes using dysprosium (III) nitrate [Dy(NO3)3] as a chemical shift reagent to resolve the signals originating from internal and external aluminium. The results showed that aluminium enters the cells at 15 min and, over a period of 4 hours, equilibrium is reached between internal and external aluminium. Citrate does not favour aluminium entering the cells at pH 5.0 and addition of EDTA extracts out all of the aluminium that has entered the cells [189]. 27Al NMR has been used to characterise the chemical forms of aluminium in xylem sap and to study the mechanism for detoxification of aluminium in roots of the tea plant (Camellia sinensis). The total concentration of aluminium in xylem sap was 0.29 mM and there were two signals in the 27Al NMR spectrum. A larger signal at 11 ppm was consistent with the peak for an aluminium-citrate model solution, suggesting that this is the main complex in which aluminium exists in xylem [190]. In roots, the quantities of aluminium and aluminium-chelating compounds (fluoride, organic acids and catechins) were measured from plants cultivated in nutrient solutions containing 0, 0.4, 1.0 and 4.0 mM aluminium at pH 4.2 for approximately 10 weeks. The levels of soluble aluminium, water-soluble oxalate and citrate, but not fluoride, malate or catechins in young roots increased with an increase in the concentration of aluminium in the treatment solution. 27Al NMR spectra of root tips and cell sap extracted from root tips that had been treated with aluminium were almost identical and had four signals, with two (11 and 16 ppm) apparently corresponding to the known chemical shifts of aluminium-oxalate complexes. In the spectra of cell sap, the resonances at 11 and 16 ppm increased with an increase in the aluminium contents. The results suggested that levels of aluminium-oxalate complexes increase in response to an increase in the aluminium level and that oxalate is a key aluminium-chelating compound in the mechanism of aluminium detoxification in the tea root [191]. Ionic aluminium is toxic for plant growth, but some plants are able to accumulate aluminium at high concentrations without showing toxicity symptoms. In this respect, 27Al NMR was used to characterise aluminium tolerance and its accumulation in tartary and wild buckwheat. Both showed high aluminium tolerance comparable to common buckwheat, they secreted oxalate rapidly from the roots in response to aluminium in a time-dependent manner and accumulated greater than 1 mg/g aluminium in the leaves after short-term exposure to aluminium. The 27Al NMR spectra revealed that aluminium was present in the form of aluminium-oxalate (1:3 ratio) in the roots and leaves, but in the form of aluminium-citrate (1:1 ratio) in the xylem sap in both species [192]. High field (19.6 T) 27Al solid-state NMR with rapid MAS (17.8 kHz) was used for an in vitro study of aluminated human brain tissue, specifically, temporal lobe tissues exposed to 0.1 mM AlCl3 and also retinal pigment epithelial cells grown in 0.1 mM AlCl3. The spectra showed multiple aluminium binding sites, good signal-to-noise ratios and apparent chemical shift dispersions (Figure 14). Tentative assignments for 27Al NMR signals in the brain tissue included those for: octahedral AlO6 (phosphate and water) (-9 to -3 ppm); condensed AlO6 units (Al-O-Al bridges) (9 ppm); tetrahedral AlO3N and/or octahedral aluminium-carbonate (24 ppm); more N-substituted aluminium and/or tetrahedral AlO4 (35 ppm) [193].
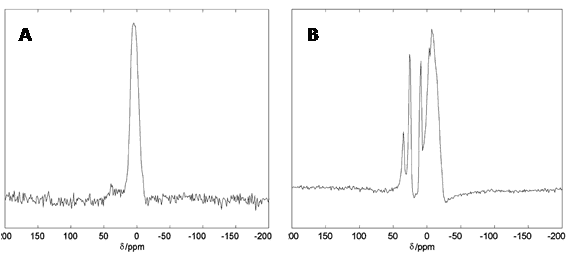
Figure 14.27Al solid-state NMR spectra of human brain tissue. 19.6 T 27Al spectra for whole nuclear extracts obtained from human retinal pigment epithelial (ARPE) cells grown in media containing 0.10 mM Al3+ (A) and total RNA isolated human brain superior temporal lobe tissues exposed to 0.10 mM Al3+ then washed in neutral saline and lyophilised (B). This Figure was reproduced with permission from Bryant et al. 2004 [193]; copyright © 2004 by Elsevier Inc.
2.12. Silicon (29Si)
The spin-1/2 nucleus 29Si (natural abundance 4.68%) produces sharp lines and has a wide chemical shift range (-346 to 173 ppm), but has low sensitivity. 29Si NMR spectra usually contain a broad background signal at around -110 ppm originating from the glass and quartz in the tube and probe, which can be suppressed by running a blank spectrum and subtracting this from the sample spectrum. 29Si NMR has very little uses with biological and biomedical samples, but one use has been for characterisation of chemical composition and silicon uptake in marine diatoms, which are key indicators of marine environmental health. 29Si MAS solid-state NMR was used for structural characterisation of biosilica deposits from four different species of diatom (Chaetoceros debilis, Chaetoceros didymum, Cylindrotheca fusiformis, Nitzschia angularis), specifically to determine the Q2:Q3:Q4 ratios. Whilst the analysis did not reveal any differences in the molecular architecture of the silica from the different diatom species, complete cells showed significantly smaller Q4:Q3 ratios (1.8-1.9) than extracted cell walls (2.5-2.8), indicating the existence of intracellular pools of less condensed silica [194]. Silicon uptake and metabolism of the marine diatom Thalassiosira pseudonana was studied by 29Si solid-state NMR and confocal laser fluorescence microscopy, especially with respect to the presence and nature of an intracellular silicon-storage pool. Diatom cells were synchronised by silicon starvation and then frozen for NMR analysis to identify potential silica precursors. NMR spectra were assigned to the various developmental stages of the dividing diatom cells and suggested that the potential silicon-storage pool consists of four-coordinated, condensed silicon [195]. 1H-29Si CP-MAS solid-state NMR was used to monitor changes in silica and organic content in the diatoms Chaetoceros muelleri and Thalassiosira pseudonana grown at three different salinities (26, 36 and 46 practical salinity units). Data showed that the Q4:Q3 area ratios of C. muelleri, grown away from standard salinities, increased in response to the formation of more condensed and/or an increase in closely associated organic matter to the Q4 component of the diatoms, which was not observed for T. pseudonana. The results suggested that there is a strong relationship between diatom composition and salinity and that C. muelleri is more sensitive to its environment than T. pseudonana [196].
2.13. Phosphorus (31P)
The spin-1/2 nucleus 31P (natural abundance 100%) has medium sensitivity, produces sharp lines and has a wide chemical shift range (-180 to 250 ppm). 31P NMR spectra are usually acquired with 1H decoupling, thus making them less crowded. The favourable properties of the 31P nucleus and its presence as a metabolite (inorganic phosphorus) and constituent of biomolecules (e.g. nucleotides, nucleic acids), phospholipid and biological membranes makes it one of the most commonly used for NMR studies with biological and biomedical systems. For example, 31P NMR can be used to probe phospholipid bilayers and biological membranes for structure, phase behaviour, dynamics and interactions with peptides, proteins, small molecules and drugs. A mention of the wide range of types of studies and samples that use 31P NMR is not possible here, so a selection of recent examples is given. 31P NMR has been used to study mitochondrial function, energy status and metabolism in resting and exercising skeletal muscle. For example, the rate of Pi to ATP exchange flux in resting muscle can be measured using saturation transfer [197] and the post-exercise recovery kinetics of pH and the concentrations of phosphocreatine, Pi and ADP contain valuable information about muscle mitochondrial function and cellular pH homeostasis in vivo [198]. Using 31P NMR it has been found that the recovery rate constant for phosphocreatine was significantly decreased in adults with Down’s syndrome compared to controls, which supports the theory of a global mitochondrial defect in Down’s syndrome [199]. Such measurements are also useful in forensic studies. Postmortem 31P NMR measurement of the α-ATP/Pi ratio in skeletal muscle showed a decrease over time from 0.445 to 0.032 over 24 hours, the method can therefore be used for determining the time of death [200]. 31P NMR has been used to measure energy status and metabolism in lesions occupying the intracranial space, where quantifying the concentrations of phosphomonoester, Pi, phosphodiester, γ-ATP, α-ATP, β-ATP with reference to phosphocreatine allowed a grading of lesions from infective to tumour [201]. 31P NMR has been used to study changes in brain bioenergetics following acute sleep deprivation. Phosphocreatine increased in grey matter after two nights of recovery sleep relative to acute sleep deprivation with no significant changes in white matter. Results also demonstrated that increases in phosphocreatine were associated with increases in electroencephalographic slow wave activity during recovery sleep. No significant changes in β-nucleoside triphosphates were observed [202]. 31P NMR analysis of phosphorus metabolites in brain regions has been used to study pathophysiology in schizophrenia [203]. In patients with gastric cancer, 31P NMR has been used to monitor premorphological alterations in gastric mucosa by measuring hypoxia levels [204]. 31P NMR measurement of the phosphomonoester/ phosphodiester ratio has been used to assess the response of chronic hepatitis C to antiviral therapy by interferon and ribavirin [205]. Other applications of solution-state 31P NMR include analysis of metabolism in dormant spores of Bacillus species by measurement of small molecules including 3-phosphoglyceric acid and ribonucleotides [206]. Also, analysis of phosphorus composition and speciation in sediments and soils, including those from Lake Erhai, southwestern China [207] and from wetland areas of eastern China [208]. 31P solid-state NMR has shown a correlation between the membrane-disruptive abilities of the β-hairpin antimicrobial peptide protegrin-1 and conformation and activity. Thus, 31P NMR lineshapes of uniaxially aligned membranes can be used as a diagnostic tool for understanding the peptide-lipid interactions of antimicrobial peptides [209]. The mechanisms of antimicrobial peptide-induced pore formation in lipid bilayers have also been investigated by oriented 31P solid-state NMR. In the cases of the peptides alamethicin and novicidin, a majority of the lipids remained in a planar bilayer conformation, but a number of lipids displayed reduced dynamics and these are involved in peptide anchoring. The results showed that alamethicin adopts a transmembrane arrangement without significant disturbance of the surrounding lipids, whilst novicidin forms toroidal pores at high concentrations and produces more extensive disturbance of the membrane [210]. 31P-19F REDOR NMR has been used to measure distances between a trifluoromethyl group and a phosphodiester in nucleic acids [211]. A 31P solid-state MRI technique has been used to observe calcification in ex vivo atherosclerotic plaques [212]. 31P solid-state NMR has been used to study the interaction of amyloid-β peptide with lipid bilayers and gangliosides. Amyloid-β strongly perturbed the structure of DMPC bilayers to form a non-lamellar phase (most likely micellar) and the ganglioside GM1 potentiated the effect of amyloid-β. The difference of the isotropic peak intensity between DMPC/amyloid-β and DMPC/GM1/amyloid-β suggested a specific interaction between amyloid-β and GM1 [213]. In an interesting application, 31P solid-state MRI has been used for in vivo visualisation of bone mineral in human wrists. Using a 3 T scanner and a quadrature wrist 31P transmit/receive coil, it was possible to obtain three-dimensional 31P images for just bone material of the wrist (Figure 15) [214].
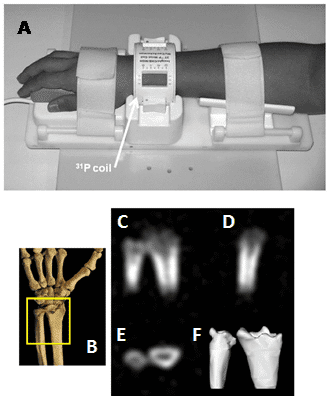
Figure 15. 31P solid-state MRI visualisation of bone mineral in human wrists. A. A 31P quadrature low pass birdcage transmit/receive coil for human wrist imaging. The enclosure containing the passive transmit/receive switch, quadrature hybrid, preamplifier and scanner interface is not shown. B-F. In vivo 31P solid-state MRI images of bone mineral in the wrist of a healthy 41 year old male: schematic view of the scanned region (A), posterior-anterior, lateral and transverse image slices (C-E), three-dimensional isosurface rendering of the full data set (F). This Figure was reproduced with permission from Wu et al. 2011 [214]; copyright © 2011 by Wiley-Liss, Inc.
2.14. Sulphur (33S)
The only NMR-active isotope of sulphur is 33S (natural abundance 0.76%), which is spin-3/2, has low sensitivity and produces very broad lines. Hence, 33S has very few uses in high-resolution NMR and there is only one published study that applies 33S NMR to biological samples. A 10 mm 33S cryogenic NMR probe was developed that operates at 9-26 K with a cold preamplifier and a cold rf switch operated at 60 K. The 33S NMR sensitivity of this cryogenic probe was up to 9.8 times greater than that of a conventional 5 mm broadband NMR probe. By application to biological samples such as human urine, bile, chondroitin sulphate and scallop tissue, it was demonstrated that the system can detect sulphur compounds having SO42- anions and –SO3– groups, but other common sulphur compounds such as cysteine were still undetectable because the 33S nuclei in these compounds are in asymmetric environments [215].
2.15. Chlorine (35Cl, 37Cl)
The two NMR-active isotopes of chlorine, 35Cl and 37Cl (natural abundance 75.6 and 24.24 %, respectively), are both spin-3/2 and produce relatively broad signals, but have a large chemical shift range (-50 to 1050 ppm). Of these, the more sensitive 35Cl is used most commonly. Early 35Cl NMR studies investigated the binding of chloride ions to zinc adenosine diphosphate complexes, zinc-pyruvate kinase complexes, carbonic anhydrases and human haemoglobin [216–219]. 35Cl NMR measurements of chloride binding to carbonmonoxy- and deoxy-dromedary hemoglobin revealed the existence of two classes of chloride-binding sites, one of high and the other of low affinity. Whilst this also resembles the situation for human hemoglobin, the number of binding sites and the association equilibrium constant for chloride binding are significantly higher in the dromedary protein. It was suggested that this difference is due to the greater number of basic residues exposed to solvent and to the higher flexibility of dromedary haemoglobin [220]. 35Cl NMR was used to reveal two new classes of chloride binding sites in the light-driven chloride pump halorhodopsin. One class exhibited low affinity (Kd much greater than 1 M) for chloride and bromide. The second class exhibited a higher affinity (Kd = 110 ± 50 mM) for chloride and also binds other anions according to the affinity series I–, SCN– > Br–, NO3- > Cl– > F–, citrate [221]. Another type of application of 35Cl NMR has been for measuring the translocation of chloride ions across membranes, which includes the co-transport of chloride into vesicles [222,223]. To assist such measurements, cobalt ions (Co2+) have been used as a chemical shift reagent to resolve the signals originating from internal and external chloride in 35Cl NMR measurements of chloride translocation with cells and vesicles [224]. The transport of chloride ions across the erythrocyte membrane has been studied by 35Cl NMR. In this case, the signal for intracellular chloride was so broad that it was virtually undetectable (linewidth greater than 200 Hz), whilst the signal for extracellular chloride was relatively narrow (linewidth around 30 Hz). Transport was totally inhibited by 4,4′-diisothiocyanostilbene-2,2′-disulphonate, which is a potent inhibitor of the erythrocyte band 3 protein [225]. A separate 35Cl and 37Cl NMR study was used to define a kinetic equation for the chloride transport cycle of band 3 in erythrocytes. The data supported a situation in which binding, dissociation and channel migration events are rapid compared to the translocation of bound chloride across the membrane. In this case, chloride binding to the transport site was described by a simple dissociation constant [Kd = Koff/Kon] rather than by a Michaelis-Menten constant [Km = (Koff + Ktranslocation)/Kon] [226]. 35Cl and 37Cl NMR relaxation measurements at various field strengths were used to study chloride binding to band 3 protein in the presence and absence of the transport inhibitor 4,4′-dinitrostilbene-2,2′-disulphonate. There were significant differences in NMR relaxation rates depending on whether the inhibitor was present or not. The results indicated that the rate of chloride association and dissociation at each external binding site occurs on a time scale of less than or equal to 5 ms, thus implying that the transmembrane flux is not limited by the rate of chloride binding to the external chloride binding site of band 3. The rotational correlation-time of chloride bound to band 3 was greater than 20 ns with a quadrupole coupling constant of approximately 2 MHz [227]. Chloride binding to band 3 in erythrocytes has also been detected by double-quantum-filtered 35Cl NMR [228]. Other applications of 35Cl and 37Cl NMR include identification of a unique pair of zinc binding sites in the human alpha 2-macroglobulin tetramer. Zinc bound at these sites did not affect the 35Cl NMR linewidth of free chloride. It was shown that additional lower affinity zinc sites exist that bind chloride weakly and cause broadening of the free chloride signal through fast exchange with bound chloride. Relaxation measurements demonstrated that chloride bound at these sites has an internal correlation time of 5.1 ns and a quadrupolar interaction of 4.2 MHz with zinc [229]. The anion binding selectivity of sarcoplasmic reticulum membranes has been studied by 35Cl NMR. Titration experiments with a series of different anions revealed that multivalent, phosphate-like anions bind much stronger to sarcoplasmic reticulum vesicles than monovalent anions like halides, whilst oxalate has an intermediate position. The binding strength decreased with decreasing ionic radius according to the following sequence: vanadate > phosphate > sulphate >> iodide > oxalate > bromide > chloride >> fluoride [230]. A feasibility study for 35Cl-MRI in humans has also been performed. It was found that 35Cl-MRI at 7 T allows in vivo imaging of 35Cl in human brain (Figure 16) and muscle in clinically feasible acquisition times (10-35 minutes) and voxel volumes (0.2-1.3 cm3). Pathophysiological changes of chloride homeostasis due to cancer or muscular ion channel disease could also be visualised [231].
Figure 16. 35Cl-MRI analysis of human brain. Transverse, sagittal, and coronal sections of a 35Cl MR imaging data set for a healthy human brain in a 67-year-old male. Two reference tubes were used (A, 103 mmol/L NaCl solution and 4% agar gel; B, 103 mmol/L NaCl solution without agar gel). An FOV of 225 x 225 mm2 is shown. A. Cl2 concentration (35Cl MR imaging) of the total tissue: The signal intensities were measured with a short TE of 0.70 ms and a long TR of 65 ms. Normalisation was performed with respect to reference tube B. Because brain tissue and reference tube B have different relaxation properties, the Cl2 concentration of brain tissue is underestimated by a factor of 1.12, when considering the measured relaxation times of brain tissue and saline solution. The Cl2 concentration was determined to be 100 mmol/L in the CSF (circle 1) and 1.12 x 32 mmol/L = 36 mmol/L in the entire tissue (circle 2). B. Differences in T1 can be taken advantage of to suppress signal from CSF (35Cl IR MR imaging). Signal intensities were normalised to reference tube A. Signal from reference tube B is well suppressed in 35Cl IR MR imaging. Colour bar = measured Cl2/Na+ concentration in mmol/L liter or measured signal intensity normalised to reference tube A. This Figure was reproduced with permission from Nagel et al. 2014 [231]; copyright © 2014 by Radiological Society of North America.
2.16. Potassium (39K)
There are three NMR-active isotopes of potassium, spin-3/2 39K (natural abundance 93.26%), spin-4 40K (natural abundance 0.01%) and spin 3/2 41K (natural abundance 6.73%), which all produce broadened signals over a relatively small chemical shift range (-30 to 35 ppm). 39K is usually preferred for NMR applications because it has the highest sensitivity and produces sharper signals than 41K. Whilst 40K produces sharper signals than 39K, it has very low sensitivity and is not really used at all. Transport of potassium ions in yeast cells and in human erythrocytes has been measured using 39K NMR with chemical shift reagents to resolve the signals originating from internal and external potassium [232,233]. Similarly, a TmDOTP shift reagent was used to assist 39K NMR measurement of intracellular potassium during ischemia in the perfused guinea pig heart [234]. A number of studies have demonstrated the usefulness of 39K NMR for in vivo measurement of potassium levels and its transport in a range of different rat tissues including blood, brain, muscle, kidney, liver, testes and mandibular salivary gland [235–238]. A number of feasibility studies have also been performed for 39K-MRI analysis of human muscle (Figure 17) and brain [239–241].
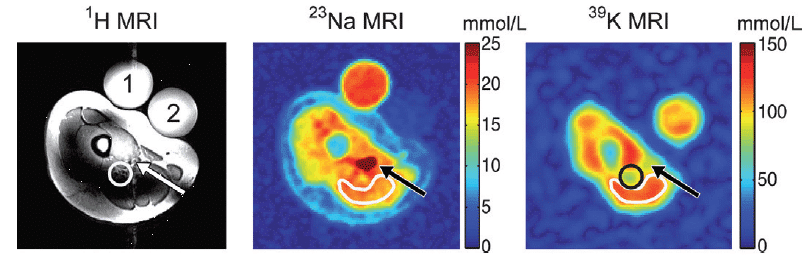
Figure 17. 39K-MRI analysis of human muscle. 1H (left), 23Na (middle) and 39K (right) MR images of the right healthy thigh muscle in a 26-year old female. 23Na and 39K MR imaging were performed with a nominal spatial resolution of 3.75 x 3.75 x 10.14 mm3 and 8 x 8 x 16 mm3, respectively. Acquisition times were approximately 30 minutes. Reference tubes containing 4% agarose gel with 20 mmol/L NaCl and 103 mmol/L KCl solutions are marked in the 1H image by 1 and 2, respectively. The femur is clearly visible on the 23Na and 39K MR images as areas of reduced signal intensity. The territory of the femoral artery and vein (arrow) exhibits high signal intensity in the 23Na MR image but reduced signal intensity in the 39K MR image. Fatty tissue that surrounds the ischiadic nerve (circles) exhibits low 39K signal intensity, while subcutaneous fat shows no 39K MR imaging signal. The mean estimated Na+ and K+ concentrations of muscle tissue (white ROI) are [Na+] = 18 mmol/L ± 1 and [K+] = 116 mmol/L ± 4. Colour bars = Measured Na+ and K+ concentrations in the 23Na and 39K MR images, respectively. This Figure was reproduced with permission from Umathum et al. 2013 [239]; copyright © 2013 by Radiological Society of North America.
2.17. Calcium (43Ca)
The NMR-active isotope of calcium is the spin-7/2 43Ca (natural abundance 0.135%), which has moderate sensitivity and produces moderately sharp signals over a relatively small chemical shift range (-35 to 35 ppm). Early 43Ca NMR experiments studied calcium binding to a range of different proteins including pancreatic prophospholipase A2 [242], calmodulin, parvalbumin and troponin C [243], pancreatic phospholipase A2 and its zymogen [244], prothrombin fragment 1 [245] bone γ-carboxyglutamic acid protein [246] and later lysosymes and alpha-lactalbumins [247]. Early 43Ca NMR experiments also studied binding of calcium to DNA [248,249]. A later 43Ca NMR study on calcium binding to DNAs involved chemical shift and linewidth measurements obtained during titration with 43CaClO4, which demonstrated the existence of at least two classes of bound 43Ca2+ ions. Binding to C. perfringens DNA (31% GC) was dominated by a delocalised, non-specific interaction. Binding to M. lysodeikticus DNA (72% GC) suggested that a small fraction of the 43Ca2+ experiences significant motional retardation and/or an increase in the electric field gradient when associated with the DNA, and therefore appeared to be locally bound to discrete sites on the DNA. Hence, the results demonstrated that higher GC content correlates with an increase in favorable Ca2+ binding environments [250]. More recently, 43Ca solid-state NMR experiments have been designed to study the structures of calcium binding sites in biological materials including metalloproteins and bone [251,252]. Indeed, natural abundance 43Ca MAS solid-state NMR has been used to obtain structural information about the coordination environment of calcium in bone using powdered bovine cortical bone samples (Figure 18) [253].
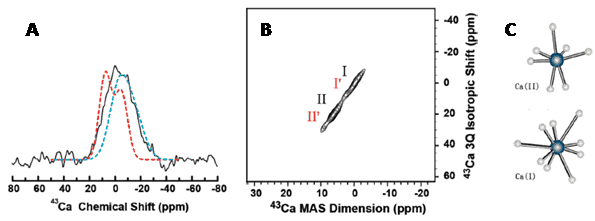
Figure 18. 43Ca MAS experiments probing the interaction of osteocalcin bone protein with calcium sites in 5% carbonated apatite. A. One-dimensional 43Ca MAS spectrum of 43Ca-enriched carbonated apatite containing osteocalcin protein at room temperature (solid black line), simulated spectrum of 5% carbonated apatite (dashed red line) and simulated spectrum of cortical bone (dashed blue line). B. Two-dimensional MQMAS spectrum correlating the 43Ca chemical shift and a triple-quantum frequency of 43Ca-enriched carbonated apatite powder sample containing osteocalcin protein at room temperature. The lengths of the excitation and conversion radio frequency pulses were 5 and 10 μs, respectively. The 2D spectrum is the resultant of 16 t1 experiments, 61,440 scans, a 0.5 s recycle delay and a 10 kHz sample spinning frequency. The 1D spectrum was obtained using a single pulse sequence with a 90° pulse length of 2 μs, 512,000 scans, a recycle delay of 250 ms and a 10 kHz sample spinning frequency. C. The two calcium coordination environments in carbonated apatite. This Figure was reproduced with permission from Xu et al. 2010 [253]; copyright © 2010 by American Chemical Society.
2.18. Vanadium (51V)
There are two NMR-active isotopes of vanadium, spin-6 50V (natural abundance 0.25%) and spin-7/2 51V (natural abundance 99.75%), which produce relatively broad signals but have a wide chemical shift range (-1900 to 0 ppm). 51V is the nucleus of choice because it is more sensitive and produces sharper signals than 50V. 51V NMR has been applied to a number of biological systems and samples.
One of the earliest applications of 51V NMR was in the study of vanadium metabolism in wild-type and respiratory-deficient strains of Saccharomyces cerevisiae [254]. 51V NMR has been used to study the binding of vanadium, vanadate and vanadium compounds to a number of different proteins including ribonuclease-T1 [255], phosphoglycerate mutase [256], chloroplast CF1-ATPase [257], transferrins [258] and prostatic acid phosphatase [259]. 51V NMR was used to study interactions of vanadate oligomers with sarcoplasmic reticulum Ca2+-ATPase. Addition of sarcoplasmic reticulum caused narrowing of the linewidth of the tetrameric vanadate signal in the presence of ATP and Ca2+, whilst there was a broadening of the signal for monomeric vanadate. Hence, ATP decreased the affinity of the enzyme for tetravanadate, whilst inducing the interaction with monomeric vanadate. In the presence of Ca2+, tetrameric and decameric vanadate were bound to sarcoplasmic reticulum ATPase, whilst monomeric vanadate only bound to sarcoplasmic reticulum when ATP was present. It was found that an increase of vesicular Ca2+ concentration counteracts monovanadate inhibition of sarcoplasmic reticulum Ca2+-ATPase activity but it does not significantly affect decavanadate inhibition [260]. 51V NMR was used to investigate the binding of vanadate to human erythrocyte ghosts. Results suggested that monomeric and polymeric vanadate species bind to the anion binding sites of band 3 protein of the erythrocyte membrane, where monomeric vanadate binds with a dissociation constant of around 0.23 mM [261]. 51V MAS solid-state NMR has been used to study the coordination environment in the 67.5-kDa protein vanadium chloroperoxidase. This was possible despite the low concentration of vanadium sites in the protein (one per molecule). Density functional theory calculations of the NMR spectra for an extensive series of active site models suggested that the vanadate cofactor is most likely to be anionic with one axial hydroxo- group and an equatorial plane consisting of one hydroxo- and two oxo- groups [262]. Correlations between 51V solid-state NMR parameters and the chemical structures of vanadium complexes have been interpreted as models for related metalloproteins. These included the isotropic chemical shift and the chemical shift anisotropy correlated with specific structural features such as the coordination number of the vanadium atom, the number of coordinating nitrogens, the number of oxygen atoms and the chemical environment of the complex. Quantitative correlations between the solid-state NMR parameters and specific bond angles and bond lengths were also obtained [263]. Further methods for analysing 51V solid-state NMR spectra of complex systems have also been developed [264].
2.19. Cobalt (59Co)
The naturally occurring isotope of cobalt, 59Co (natural abundance 100%), is NMR-active with a spin of 5/2. 59Co has high sensitivity and produces broad signals, but it does have an extremely wide chemical shift range (-4000 to 14000 ppm). 59Co NMR has found some applications with biological and biomedical systems and samples. A method using 59Co NMR was developed for measuring the volume of intracellular water space. This is based on the signal for an inert, stable and membrane-impermeable cobalt(III) compound such as [Co(CN)6]3− or [Co(imidazole)6]3+. In its first application, variation of the intracellular water space in human erythrocytes as a function of osmolality was measured [265]. The method was later used in studies of experimental acute pancreatitis, whereby interstitial and total water volumes of the pancreas were estimated by measuring the distribution of [Co(CN)6]3−. In the healthy pancreas, the interstitial compartment comprised 35% of the tissue volume. In the diseased pancreas, the penetration volume of [Co(CN)6]3− reached 90% of the total tissue volume, which was indicative of extensive membrane injury [266]. The method has been used for measuring intracellular water volumes in a range of different cell suspensions and perfused organs [267]. 59Co NMR has been used for in vivo thermometry with liposomes containing cobalt complexes, which is useful for making localised temperature measurements in tissue during hyperthermia treatment of cancer. The method uses the complex tris(ethylenediamine) cobalt(III) trichloride as a temperature sensor by determining the temperature dependence of its 59Co NMR chemical shift (Figure 19). Encapsulation within liposomes allows targeting of the agent to the reticuloendothelial system and temperature changes in the order of 0.1 ºC have been measured in vivo in rats [268].
Figure 19. 59Co NMR in vivo thermometry with liposomes containing a cobalt complex. A. Three representative 59Co spectra showing the change in chemical shift as a function of temperature. B. Resonant frequency shift of the cobalt (III) nucleus in the Co(en)3C13 complex as a function of temperature for a solution in water. The standard error for temperature measurements using the Luxtron fibre-optic thermometer was 0.1 ºC and the standard error for the measured frequency shift was also 0.1 ºC. This Figure was reproduced with permission from Webb et al. 1995 [268]; copyright © 1995 by Taylor & Francis.
59Co NMR has been used to probe interactions of [Co(NH3)6]3+ with helical B-DNA (Braunlin et al., 1987) and for chiral recognition of deoxyoligonucleotides by δ- and λ-tris(ethylenediamine)cobalt(III) [269]. 59Co solid-state NMR has also been used to probe metal binding sites in polynucleotides. This included elucidation of magnesium-nucleic acid binding sites by using [Co(NH3)6]3+ as surrogate for [Mg(H2O)6]2+ [270].
2.20. Copper (65Cu)
The two NMR-active isotopes of copper, 63Cu (natural abundance 69.15%) and 65Cu (natural abundance 30.85%), are both spin-3/2 and produce broad signals, but have a large chemical shift range (-300 to 800 ppm). 63Cu is the more sensitive nucleus, whilst 65Cu produces slightly narrower signals. There are very few applications of 63Cu or 65Cu with biological samples. Low temperature 65Cu NMR measurements have been performed on the blue copper protein azurin in the reduced copper I state. Measurements at18.8 T and 10 K produced a strongly second order quadrupole perturbed spectrum, which gave a 65Cu quadrupole coupling constant of ± 71.2 ± 1 MHz, corresponding to an electric field gradient of ± 1.49 atomic units at the copper site, and an asymmetry parameter of approximately 0.2 [271].
2.21. Zinc (67Zn)
The only NMR-active isotope of zinc is spin-5/2 67Zn (natural abundance 4.102%), which is low sensitivity and produces moderately broad signals. 67Zn NMR applications with biological samples are scarce. 67Zn solid-state NMR has been used to study the minimal DNA binding domain of human nucleotide excision repair protein XPA [272] and to investigate the zinc binding site in the zinc protein rubredoxin [273]. Residue ionisation in the zinc-dependent deacetylase of bacterial lipid A synthesis (LpxC) has been directly observed by 67Zn solid-state NMR. The pH-dependence of 67Zn solid-state NMR lineshapes were measured for both wild-type and mutant (H265A) forms of Aquifex aeolicus LpxC, each in the absence of substrate (resting state). The spectrum of wild-type LpxC at pH 6 contained two overlapping quadrupole lineshapes with C q values of 10 and 12.9 MHz, whilst the spectrum for a sample prepared at pH 9 was dominated by the appearance of a third species with a C q of 14.3 MHz. Spectra of the mutant were pH-independent, where a C q of 9.55 MHz was sufficient to describe both low and high pH data. Quantum mechanical/molecular mechanical modelling of the H265A mutant suggested that over this pH range water is bound to the zinc ion whilst Glu78 is protonated (Figure 20) [274].
Figure 20. Residue ionisation in Aquifex aeolicus LpxC observed by 67Zn solid-state NMR. QM/MM optimised quantum regions of WT LpxC with doubly protonated His265, water bound to zinc and a proton added to Glu78 Oε1 and Oε2 in (A) and (B), respectively, and H265A LpxC with water bound to zinc and (C) Glu78 Oε1H and (D) Glu78 Oε2H. This Figure was reproduced with permission from Lipton et al. 2008 [274]; copyright © 2008 by Journal of the American Chemical Society.
2.22. Selenium (77Se)
The NMR-active isotope of selenium, spin-1/2 77Se (natural abundance (7.63%), has low sensitivity but produces narrow signals over a very wide chemical shift range (-1000 to 2000 ppm). 77Se NMR applications with biological samples are relatively scarce. An early study demonstrated the feasibility of using 77Se NMR for observing selenium covalently attached to proteins. The disulphide bonds of ribonuclease-A and lysosyme were reductively cleaved under denaturing conditions and the resulting sulphydryl groups were treated with the reagent 6,6′-diselenobis(3-nitrobenzoic acid) to give proteins containing covalently attached selenium in the form of selenenyl sulphides. In high-resolution 77Se NMR spectra of these proteins under denaturing conditions, five to six signals within a chemical shift range of 14-15 ppm were observed for each protein and were compared to the chemical shifts of several model selenenyl sulphides derived from cysteine [275]. A later study investigated the NMR relaxation properties of 77Se-labelled proteins. Cysteine residues in the proteins bovine hemoglobin, reduced ribonuclease A and glutathione were labelled with 77Se by reaction with [77Se]6,6′-diselenobis(3-nitrobenzoic acid) and the resultant species contained Se-S linkages with 77Se NMR signals in the range 568-580 ppm. It was concluded that when there are no complications from protein aggregation or chemical exchange, the chemical shift anisotropy values anticipated to exist in selenoproteins should result in signals with linewidths in the range 27 to 170 Hz, depending on field strength. Such signals should therefore be observable in the intact protein, if 77Se-enriched material is available [276]. 77Se NMR was used to characterise 77Se-labelled ovine erythrocyte glutathione peroxidise. Lambs, maintained on a selenium-deficient diet supplemented with 94 atom % Na277SeO3, were used as the source of 77Se-enriched erythrocyte glutathione peroxidise, for which the content of 77Se reached 88%. Monthly bleeding of two animals produced approximately 20 mg of 77Se-enriched glutathione peroxidase in pure form [277]. More recently, 77Se NMR has been used for site-specific pKa determination of selenocysteine residues in seleno-vasopressin [278].
2.23. Bromine (79Br, 81Br)
The two naturally occurring isotopes of bromine, 79Br (natural abundance 50.69%) and 81Br (natural abundance 49.31%), are both NMR-active with spin-3/2 and produce broad signals, but have a wide chemical shift range (-500 to 100 ppm). 81Br is the more sensitive nucleus and produces slightly narrower signals than 79Br. Because 79Br has a frequency very similar to that of 13C, is observable using a carbon specific probe and produces clear quadrupolar spinning sidebands in the solid-state, it is widely used in MAS solid-state NMR for adjusting the magic angle. 79Br and 81Br NMR applications with biological samples are very scarce. An early study used 79Br and 81Br NMR to probe the reversible association of α-chymotrypsin and a mercury-containing substrate (4-bromomercuriocinnamic acid, BrHgCin), which rapidly exchanges bromide ions. It was found that the rate of bromide exchange is diffusion-limited and faster than the rate of reorientation of the BrHgCin-α-chymotrypsin complex. The rapid rate of bromide exchange with the complex was not compatible with the side chain of BrHgCin being entirely buried in a nonpolar pocket on the enzyme but compatible with the side chain being exposed to the solution [279].
2.24. Krypton (83Kr)
The NMR-active isotope of krypton, spin-9/2 83Kr (natural abundance 11.50%), has low sensitivity but produces narrow signals over a chemical shift range of 40 to 180 ppm. The main use of 83Kr has been as a contrast agent for 83Kr-MRI analysis of lungs. The feasibility for using hyperpolarised 83Kr for imaging was first tested in canine lung tissue by using krypton gas with natural abundance isotopic distribution [280]. 83Kr-MRI has been used for ex vivo imaging of rat lungs using natural abundance krypton gas [281] and to detect tobacco smoke deposition on a model glass surface coated with bovine lung surfactant extract [282]. A recent study used 83Kr-MRI with a surface quadrupolar relaxation (SQUARE) contrast method for imaging of alveolar degradation in a rat model of emphysema [283].
2.25. Rubidium (87Rb)
There are two NMR-active isotopes of rubidium, spin-5/2 85Rb (natural abundance 72.17%) and spin-3/2 87Rb (natural abundance 27.83%), which produce relatively broad signals in the chemical shift range -80 to 30 ppm. 87Rb has the highest sensitivity and produces less broad signals than 85Rb. The main applications of 87Rb NMR originate from the fact that rubidium can be used to replace potassium in biological systems and 87Rb is more sensitive than 39K. Indeed, 87Rb NMR measurements in human erythrocytes, using the chemical shift reagent dysprosium(III) triethylenetraamine-N,N,N’,N”,N”‘,N”‘-hexaacetic acid (DyTTHA3-) to resolve internal and external signals, demonstrated that rubidium transport proceeds via Na+,K+-ATPase as there was inhibition of uptake in presence of ouabain [284]. Both 87Rb NMR and 87Rb-MRI have been used for studies of potassium transport and energetics in mammalian cells, organs and in vivo. This includes studies on isolated perfused rat and pig hearts and also on kidney, skeletal muscle, salivary gland and red blood cells. The method has been used for measuring the kinetics of unidirectional rubidium uptake and efflux and steady-state rubidium levels [285]. For example, 3D 87Rb-MRI analysis of isolated pig hearts allowed the effects of regional ischemia on rubidium uptake to be investigated. In the control group, distribution of rubidium in the left ventricle and the intensities of the three-dimensional 87Rb images were uniform. In the ischemic group, images showed a reduced 87Rb intensity, corresponding to decreased rubidium content (33 ± 11% of normal) [286]. 87Rb NMR was used to measure mitochondrial potassium fluxes in whole rat hearts [287] and potassium transport in mouse hearts [288]. 87Rb-MRI has been used to investigate ischemia and infarction in blood-perfused pig hearts [289] and to measure potassium transport and energetics in Kir6.2 channel knockout mouse hearts [290].
2.26. Cadmium (111Cd, 113Cd)
There are two NMR-active isotopes of cadmium, 111Cd (natural abundance 12.80%) and 113Cd (natural abundance 12.22%), which are both spin-1/2 and produce narrow signals over a wide chemical shift range (-650 to 0 ppm). 113Cd is the more commonly used nucleus because it is slightly more sensitive than 111Cd. A large majority of the applications of 111Cd and 113Cd NMR have been for probing the binding properties and structures of metal binding sites in metalloproteins and in proteins that have metals as ligands or substrates, including metal transporters. The relatively few applications of 111Cd NMR include a study of the binding of novel N-hydroxybenzene-sulphonamide carbonic anhydrase inhibitors to native and 111Cd-substituted carbonic anhydrase [291] and characterisation of zinc(II) binding to the peptide amyloid-β1-16 linked to Alzheimer’s disease [292]. There are a number of 111Cd NMR studies with metallothioneins, which are small (6-7 kDa) cysteine- and metal-rich proteins. The domain specificity of silver and copper binding to metallothionein was studied by 111Cd NMR. For example, titration of 111Cd7MT with silver indicated that silver ions bind preferentially to the β-domain of the protein to form the metal hybrid species (Cd4)α(Ag6)βMT. Once the β-domain is filled, additional silver ions displace cadmium from the α-domain to form (Ag6)α(Ag6)βMT. The metal displacement reaction is cooperative and the two domains react independently of one another. Copper reacts with Cd7MT in a manner similar to silver, except that, unlike silver, copper appears to produce intermediate species that may contain mixed-metal clusters. The differential affinities of the two domains for monovalent and divalent metal ions along the availability of facile pathways for metal exchange may be features that enable metallothionein to function simultaneously in the metabolism of different metal ions [293]. Whilst it was already known that in mammalian metallothionein, zinc is exclusively coordinated with Cys-thiolate to form clusters in which the metal is thermodynamically stable but kinetically labile, little was known about coordination in prokaryotic metallothionein. Hence, 111Cd NMR measurements with a prokaryotic maetallothionein showed how metal coordination involves the imidazole moieties of histidine residues [294]. 111Cd NMR was also used to demonstrate histidine coordination in a plant zinc-metallothionein. Two conserved histidine residues participated in metal binding in a novel cluster composition that was different compared to those from mammals or cyanobacteria [295]. 113Cd NMR has been used in a large number of studies with metallothioneins, including correlation of the 113Cd chemical shift with the nature of the coordinating ligands (N, O, S) and coordination number/geometry and structure determination of the metal binding sites and whole proteins [296,297]. 113Cd NMR was used to determine the structures of the multiple metal-binding sites in the two major isoproteins of metallothionein from mammalian livers (rabbits, calf and human) and from giant mud crab hepatopancreas. This was achieved by metal replacement with 113Cd and then analysis of 113Cd NMR spectra to provide structures for the binding site clusters [298]. Two-dimensional heteronuclear 1H-113Cd correlated spectroscopy was used for obtaining through-bond connectivities between the metals and sequentially assigned cysteine residues to assist spatial structure determination of rabbit liver metallothionein-2. The protein comprised two domains enclosing a three-metal cluster and a four-metal cluster. The metal coordination within these clusters was different from that of (Cd5, Zn2)-MT-2 from rat liver as determined by X-ray crystallography. In the two structures different metal-cysteine combinations were apparent for 5 of the 12 connectivities in the three-metal cluster and for 14 of the 16 connectivities in the four-metal cluster [299]. Chinese hamster metallothioneins with mutations in invariant cysteine residues had altered metal binding capacity, cadmium resistance and 113Cd NMR spectra. Analysis of the 113Cd NMR spectra for the mutant metallothioneins provided an indication as to the structural basis for the effects of each mutation on metal binding [300]. The three-dimensional structure of metallothionein-1 from blue crab was determined by solution-state NMR, including heteronuclear 1H-113Cd correlation experiments. The protein binds six divalent metal ions in two separate metal-binding clusters, which reside in two distinct domains. Structure determination involved measurement of 24 metal-to-cysteine connectivities from the 1H-113Cd correlation experiments. The only element of regular secondary structure in either of the two metal binding domains was a short segment of helix in the C-terminal α-domain between Lys42 and Thr48. The folding of the polypeptide backbone chain in each domain, however, produced several type I beta turns [301]. The effect of nitric oxide on metal release from metallothionein-3 [302] and the reaction of human metallothionein-3 with the platinum-based anticancer drugs cisplatin and transplatin [303] have also been investigated by 113Cd NMR. 113Cd NMR has been used in applications with a large number of other types of proteins. One of the earliest studied the effect of pH, bicarbonate and cyanide on carbonic anhydrases [304]. Other examples are studies of the metal binding sites in blue copper proteins [305], a yeast superoxide dismutase [306], human retinoic acid receptor-β DNA binding domain [307], a bacterial phosphotriesterase [308] , bovine and human α-lactalbumin and equine lysozyme [309] and cabbage histidinol dehydrogenase [310]. 113Cd NMR was also used to dentify the coordinating residues in the DNA binding domain of the glucocorticoid receptor [311]. The forms of cadmium in the leaves of Thlaspi caerulescens and in xylem sap of Arabidopsis halleri, which are both cadmium hyperaccumulators, have been identified by 113Cd NMR. In the former case it was a cadmium-malate complex [312] and in the latter case it was Cd(NO3)2 [313]. 113Cd NMR has been used to study interactions between cadmium(II) and extracellular organic matter released by the green alga Selenastrum capricornutum [314] and to characterise interactions between cadmium and models of the main components of soil organic matter, specifically exopolysaccharides, exudates of roots (polygalacturonic acid) and soil bacteria [315]. 113Cd solid-state NMR has been used to probe metal binding to the Escherichia coli zinc exporter ZitB with amplified expression in native membranes. Direct observation of 113Cd binding to ZitB allowed competitive titrations with a range of other metals. These indicated that ZitB is able to bind both nickel and copper, as well as zinc [316].
2.27. Iodine (127I)
The only naturally occurring isotope of iodine, 127I, is NMR-active with a spin of 5/2 and has medium to high sensitivity. Whilst 127I has a very wide chemical shift range (-100 to 4100 ppm), it also tends to produce very broad signals. Hence, applications of 127I NMR with biological systems and samples are very scarce. 127I NMR was used to study the binding of iodide by lactoperoxidase. The 127I NMR signal for iodide binding showed no competition with cyanide and spectra revealed that the binding of iodide is facilitated by protonation of an ionisable group with a pKa value of 6.0-6.8, which was presumed the distal histidyl residue [317]. 127I NMR was also used to study iodide binding to Arthromyces ramosus peroxidise. This showed that the binding of iodide depends on protonation of an amino acid residue with a pKa of around 5.3, which was presumed to be the distal histidine (His56), which is 7.8 Å away from the iodide ion [318].
2.28. Xenon (129Xe)
There are two NMR-active isotopes of xenon, spin-1/2 129Xe (natural abundance 26.40%) and spin-3/2 131Xe (natural abundance 21.23%), which have low to medium sensitivity and a very wide chemical shift range (-5700 to 100 ppm). 129Xe produces the sharpest signals and is used most commonly. The hyperpolarisation of 129Xe gas has allowed a large number of NMR applications with biological and biomedical systems and samples [319]. The two main approaches that have emerged are 129Xe NMR-based biosensors and 129Xe-MRI. Xenon-encapsulating molecules called cryptophanes emerged as probes for 129Xe NMR that could be used in biosensors. The 1.70 Å resolution crystal structure of a cryptophane-derivatised benezenesulphonamide complexed with human carbonic anhydrase II showed how an encapsulated 129Xe atom can be directed to a specific biological target. The structure indicated that the cryptophane cage does not strongly interact with the surface of the protein, which may enhance the sensitivity of 129Xe NMR spectroscopic measurements in solution (Figure 21) [320]. More recently, the (+) and (-) enantiomers for a cryptophane-7-bond-linker-benzenesulphonamide biosensor (C7B) have been synthesised and biosensor binding to carbonic anhydrase II was characterised for both enantiomers by hyperpolarised 129Xe NMR [321]. A biosensor based on the non-selective grafting of cryptophane precursors on holo-transferrin have been assessed, where cell uptake of the sensor was accompanied by hyperpolarised 129Xe NMR detection [322].
Figure 21. Cryptophanes as xenon-encapsulating molecules as probes for a 129Xe NMR biosensor of carbonic anhydrase II.
A. MoMo and PoPo enantiomers of the cryptophane-A-derived carbonic anhydrase biosensor. The benzenesulphonamide moiety serves as an affinity tag that targets the Zn2+ ion and the R1 substituents contain triazole propionate moieties that enhance aqueous solubility. B. Stereoview of a simulated annealing omit map showing 1-MoMo (blue) and 1-PoPo (red) bound in the active site (1.9 σ contour, teal). A Bijvoet difference Fourier map (2.0 σ, black) confirms encapsulation of Xe (yellow). This Figure was reproduced with permission from Aaron et al. 2008 [320]; copyright © 2008 by American Chemical Society.
Targeting and delivery of a peptide-modified cryptophane to cells expressing αvβ3 integrin receptor, which is overexpressed in many human cancers, was reported for a 129Xe NMR biosensor. Cryptophane was functionalised with cyclic RGDyK peptide and 129Xe NMR revealed a single resonance at 67 ppm for the 129Xe-cryptophane-cyclic RGDyK biosensor. Introduction of αIIbβ3 integrin receptor in detergent solution generated a new ‘bound’ 129Xe biosensor signal that was shifted 4 ppm downfield from the ‘free’ 129Xe biosensor [323]. A ‘clickable’ and highly water-soluble PEGylated cryptophane has been developed as a potential universal platform for 129Xe NMR biosensors. The molecule is easily functionalised by Huisgen cycloaddition and exhibits excellent xenon-encapsulation properties (Figure 22) [324]. Molecular imaging of cancer cells using a bacteriophage-based 129Xe NMR biosensor has been achieved [325] and an antibody-based, modular biosensor for 129Xe NMR molecular imaging of cells has been developed using cryptophanes as the chemical host for hyperpolarised nuclei [326]. Hyperpolarised 129Xe chemical exchange saturation transfer (Hyper-CEST) NMR techniques allow the ultrasensitive (1 picomolar) detection of xenon in cryptophane host molecules. Hyper-CEST 129Xe NMR has been used to detect Bacillus anthracis and Bacillus subtilis spores in solution and to interrogate the layers that comprise their structures. 129Xe-spore samples were selectively irradiated with radiofrequency pulses, the depolarised 129Xe was returned to aqueous solution and depleted the 129Xe-water signal, which provided a measurable contrast. Removal of the outermost spore layers in B. anthracis and B. subtilis (the exosporium and coat, respectively) enhanced 129Xe exchange with the spore interior [327].
Figure 22. ‘Clickable’ and highly water-soluble PEGylated cryptophanes for 129Xe NMR biosensors. 129Xe NMR spectra obtained at 11.7 T and 293 K for compound 1 (9 mm) in D2O (A) and compound 11 (37 mm) in D2O (B). Inset: subspectra obtained by a succession of 32 selective Gaussian pulses of 500 ms centered at the Xe@cryptophane frequency and acquisitions with a 50 ms repetition time. This Figure was reproduced with permission from Delacour et al. 2013 [324]; copyright © 2013 by WILEY-VCH Verlag GmbH & Co. KGaA, Weinheim.
Hyper-CEST 129Xe NMR has also been used for the simultaneous detection of lead, zinc and cadmium ions at nanomolar concentration [328]. An alternative method called ultra-fast Z-spectroscopy has been developed as a powerful means to detect low concentrations of 129Xe NMR-based sensors and to measure the in-out xenon exchange [329]. Furthermore, optical hyperpolarisation and NMR detection of 129Xe has been achieved on a microfluidic chip (Figure 23). The chip was capable of 129Xe polarisations of greater than 0.5% at flow rates of several microlitres per second, compatible with typical microfluidic applications. In situ optical magnetometry was employed to sensitively detect and characterise the 129Xe polarisation at magnetic fields of 1 μT. Because the device was constructed using standard microfabrication methods, it may enable implementation of highly sensitive 129Xe NMR in compact, low-cost, portable devices [330].
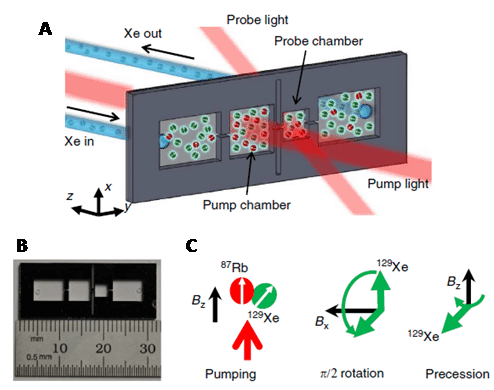
Figure 23. A microfluidic chip 129Xe polariser. A. A gas mixture containing 400 Torr N2 and 200 Torr Xe in natural isotopic abundance (26.4% 129Xe content) flows from a bulk gas manifold into the inlet chamber, through the pump and probe chambers and out of the outlet chamber. The chip is loaded with 2 mg of 87Rb metal. Unpolarised 129Xe atoms entering the pump chamber become polarised through spin exchange with optically pumped 87Rb. The 129Xe then moves downstream, passes through a microchannel into the probe chamber and eventually exits the device through the output chamber. Optical characterisation of the 129Xe polarisation in the pump and probe chambers is carried out using the ensemble of 87Rb atoms in each chamber as in situ magnetometers. B. The silicon chip footprint is 3 cm x 1 cm, with a thickness of 1 mm. The dimensions of the pump and probe chambers are 5 mm x 5 mm x 1 mm and 3 mm x 3 mm x 1 mm, respectively, whereas the channel connecting the pump and probe chambers is 1 mm x 0.3 mm x 0.3 mm. Two tall, narrow grooves are etched from the middle of the chip to provide thermal isolation between the two sides of the device. C. Pumping and probing sequence for 129Xe. Continuous pumping is carried out in presence of a longitudinal field Bz = 0.8 mT. Every 10-20 s, a transverse DC field of magnitude 5.3 mT is switched on for 4 ms to tip the 129Xe atoms onto the x-y plane and initiate 129Xe precession about the longitudinal axis. This Figure was reproduced with permission from Jiménez-Martínez et al. 2014 [330]; copyright © 2014 by Nature Publishing Group.
Because of the generally inert nature of xenon gas and the absence of a xenon background in biological tissues, hyperpolarised 129Xe-MRI has emerged as a viable clinical technique, especially for imaging of lung function and associated conditions such as asthma and COPD. Indeed, studies in humans using techniques for measuring ventilation, diffusion and partial pressure of oxygen have demonstrated results for hyperpolarised 129Xe-MRI comparable to those obtained using hyperpolarised 3He-MRI, even though 129Xe produces reduced signals due to its smaller magnetic moment. Xenon also has the advantage of readily dissolving in lung tissue and blood following inhalation, which makes hyperpolarised 129Xe particularly attractive for investigating some characteristics of lung function, such as gas exchange and uptake, which cannot be accessed using 3He [331]. A quantitative comparison of hyperpolarised 3He- and 129Xe-MRI in healthy individuals and patients with COPD was performed, in which images were obtained within five minutes of each other. In patients with COPD, ventilation defect percentages obtained with 129Xe were significantly greater than those obtained with 3He. This suggested incomplete or delayed filling of lung regions that may be related to the different properties of 129Xe gas and physiologic and/or anatomic abnormalities in COPD [332]. Human lung structure and physiology in healthy individuals were quantified by hyperpolarised 129Xe-MRI. Values obtained from xenon uptake (averaged over all individuals) included: surface-area-to-volume ratio (210 ± 50 cm-1); total septal wall thickness (9.2 ± 6.5 μm); blood-air barrier thickness (1.0 ± 0.3 μm); hematocrit (27 ± 4%); pulmonary capillary blood transit time (1.3 ± 0.3 s). All were in good agreement with literature values from invasive experiments [333]. A regional mapping study of gas uptake by blood and tissues (lung parenchyma and plasma) in the human lung was performed using hyperpolarised 129Xe-MRI. A three-dimensional, multi-echo, radial-trajectory pulse sequence was used to obtain ventilation (gaseous 129Xe), tissue and red blood cell images in healthy subjects, smokers and asthmatics. Signal ratios (total dissolved 129Xe to gas, tissue-to-gas, red blood cell-to-gas and red blood cell-to-tissue) were calculated from the images. Healthy subjects demonstrated generally uniform values within coronal slices and a gradient in values along the anterior-to-posterior direction (Figure 24). In contrast, images and associated ratio maps in smokers and asthmatics were generally heterogeneous and exhibited values mostly lower than those in healthy subjects. Whole-lung values of total dissolved 129Xe-to-gas, tissue-to-gas, and red blood cell-to-gas ratios in healthy subjects were significantly larger than those in diseased subjects [334].
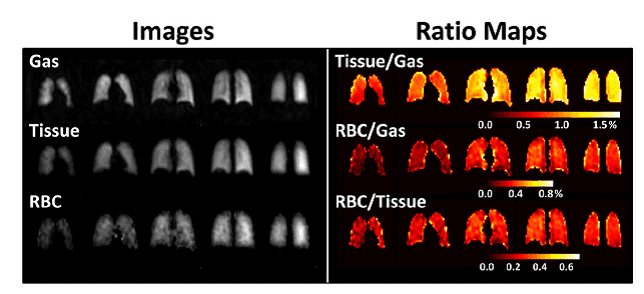
Figure 24. Hyperpolarised 129Xe-MRI mapping of human lung. Coronal gas, tissue and RBC images (left) and corresponding tissue-to-gas, RBC-to-gas and RBC-to-tissue ratio maps (right) of whole lung from a healthy individual. While dissolved-phase signal intensities and ratio values were generally uniform within each coronal slice, the tissue and RBC images, as well as tissue-to-gas and RBC-to-gas ratio maps, showed an anterior-to-posterior gradient associated with the gravity-dependent gradient in lung tissue density in the supine position. This Figure was reproduced with permission from Qing et al. 2014 [334]; copyright © 2013 by Wiley Periodicals, Inc.
Hyperpolarised 129Xe-MRI has also been used for the imaging of other organs and tissues. Using a rat permanent right middle cerebral artery occlusion model, it was demonstrated that hyperpolarised 129Xe-MRI can be used for the in vivo detection of the hypoperfused area of focal cerebral ischemia [335]. Another study used hyperpolarised 129Xe-MRI for detection of brown adipose tissue (BAT) and thermogenic activity in mice. There was a greater than 15-fold increase in xenon uptake by BAT during stimulation of BAT thermogenesis, which enabled acquisition of background-free maps of BAT in both lean and obese mice (Figure 25).
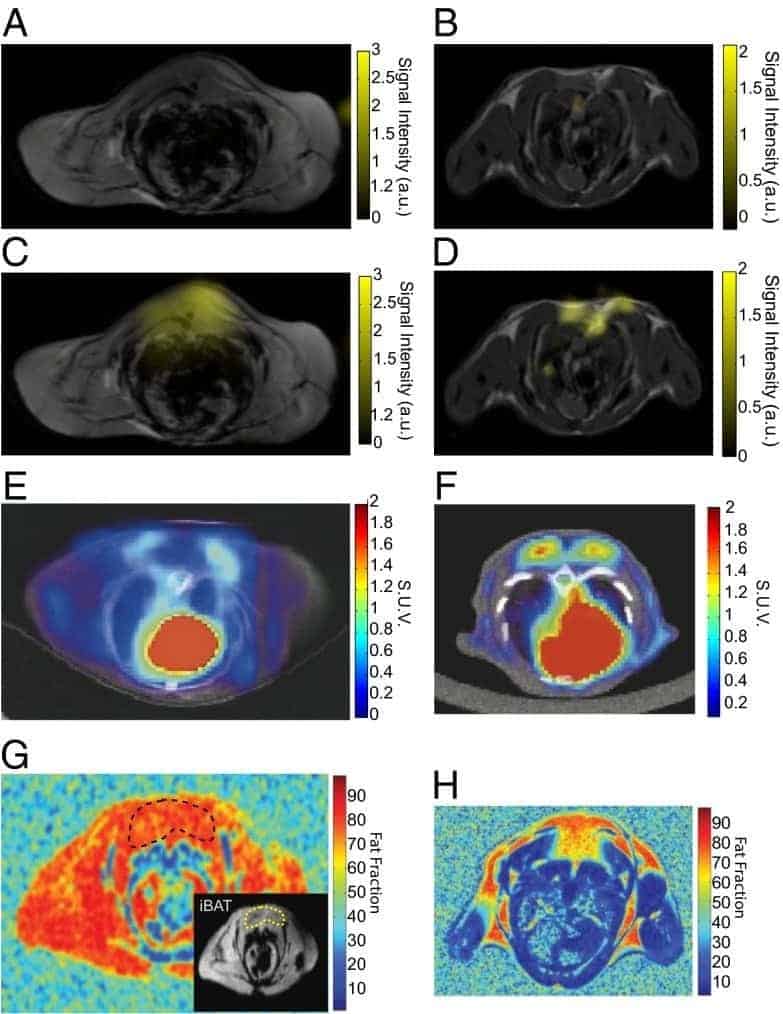
Figure 25. Hyperpolarised 129Xe-MRI maps of interscapular BAT in lean and obese mice and comparison with [18F]FDG-PET/CT. The figure shows detection of BAT by hyperpolarised 129Xe-MRI, [18FDG]-PET, and fat fraction 1H-MRI in lean (right) and obese (left) mice. Hyperpolarised dissolved-phase xenon images are displayed as a false colour overlay on the corresponding 1-mm-thick axial proton reference image at baseline (A and B) and during stimulation of BAT thermogenesis (C and D) in the same animals. E and F. [18F]FDG-PET/CT images in a lean (F) and obese (E) animal during stimulation of BAT thermogenesis. G and H. Fat fraction 1H-MRI measurements in a lean (H) and obese (G) mouse. The inset in G delineates interscapular BAT in the obese mouse. This Figure was reproduced with permission from Branca et al. 2014 [336]; copyright © 2014 by Branca et al.
The study also demonstrated in vivo MRI thermometry of BAT by hyperpolarised 129Xe gas and the linear temperature dependence of the chemical shift of xenon dissolved in adipose tissue was used to directly measure BAT temperature and to track in vivo thermogenic activity [336]. Because inhaled xenon atoms are transferred from the lung via the bloodstream to the brain, this offers an opportunity to image the brain using hyperpolarised 129Xe-MRI. Also, the longitudinal relaxation time (T1) of hyper-polarised 129Xe is inversely proportional to the pulmonary oxygen concentration in the lung because oxygen molecules are paramagnetic. In contrast, the T1 of 129Xe is proportional to the pulmonary oxygen concentration in the blood, because the higher pulmonary oxygen concentration will result in a higher concentration of diamagnetic oxyhemoglobin. Hence, there should be an optimal pulmonary oxygen concentration for a given quantity of hyperpolarised 129Xe in the brain. These relationships have been explored in theoretical and in vivo experiments for improving the 129Xe signal in brain (Figure 26) [337].
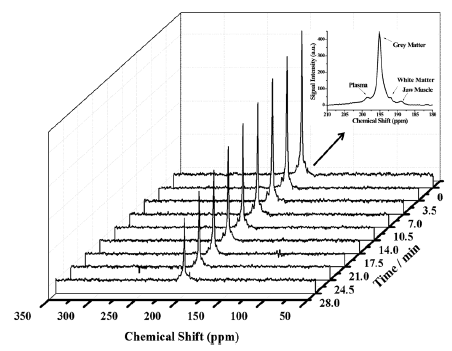
Figure 26. Typical dynamic spectra of the hyperpolarised 129Xe-MRI signal in rat brain. The hyperpolarised 129Xe signal decreased with increasing interval time. The inset shows assignments of the visible dissolved 129Xe peaks in the brain, and the signals at 189 ppm and 195 ppm originate from the jaw muscle and grey matter, respectively. The other signals at 192 ppm and 197 ppm likely originate from the white matter and plasma, respectively. This Figure was reproduced with permission from Li et al. 2016 [337]; copyright © 2016 by John Wiley & Sons, Ltd.
2.29. Caesium (133Cs)
The only naturally occurring isotope of caesium, 133Cs, is NMR-active with a spin number 7/2. 133Cs has medium sensitivity and produces relatively narrow signals over a chemical shift range of -30 to 130 ppm. The main applications of 133Cs NMR wih biological and biomedical systems and samples are based on the fact that caesium can be used as a substitute for potassium [338]. Indeed, caesium is accumulated in the intracellular space primarily through the action of the Na+-K+-ATPase and dietary loading of 133Cs can be used as a potassium substitute in NMR studies of tissues [339]. An early study used 133Cs NMR to measure uptake of caesium ions by human erythrocytes and perfused rat heart. Spectra exhibited two sharp resonances originating from intra- and extracellular caesium, separated by a chemical shift of 1.0-1.4 ppm. Hence, resonances can be resolved without the addition of paramagnetic shift reagents that are required to resolve resonances of the other metal ions. The uptake of caesium ions by erythrocytes occurred at approximately one-third of the reported rate for potassium ions and was reduced by a factor of 2 upon addition of ouabain [340]. 133Cs NMR was also used to compare caesium uptake into human erythrocytes from abstemious individuals and alcoholic patients, but no discernible differences were observed [341]. 133Cs NMR was used as an in vivo probe for studying subcellular compartmentation and ion uptake in maize root tissue. Three 133Cs-NMR signals were observed in spectra of CsCl-perfused and CsCl-grown maize seedling root tips. Two relatively broad lower field resonances were assigned to the subcellular, compartmented caesium ions in the cytoplasm and vacuole, respectively. 133Cs NMR spectra of excised, maize root tips and excised top sections of the root adjacent to the kernel, each grown in 10 mM CsCl showed a difference in the relative areas of the resonance corresponding to the distinct cytoplasm/vacuole volume ratio of the different sections of the root. The results suggested that 133Cs NMR might be useful for studying ionic strength and osmotic pressure associated chemical shifts and transport properties of caesium ions, and hence potassium ions, in subcellular compartments of plant tissues [342]. Using both excised and in situ tissues from rats fed and administered CsCl, it was demonstrated how 133Cs NMR can be used as a probe of intracellular space. Chemical shifts, relaxation properties, sensitivity and detectability of caesium in tissues were investigated. Two 133Cs NMR resonances, representing intra- and extracellular caesium, were detected in blood, whilst one resonance was detected in brain, kidney and muscle tissue [343]. Studies of ion transport in perfused and septic rat heart have been performed using 133Cs as an NMR-active substitute for potassium [344,345]. 133Cs NMR measurements with porcine aortic endothelial cells have demonstrated that nitric oxide radicals, through stimulation of guanylate cyclase, cause a reduction Na+,K+-ATPase activity [346]. 133Cs NMR measurements on isolated perfused hepatocytes from caesium-fed rats demonstrated two distinct intracellular environments, evident as compartments with different 133Cs chemical shifts and containing different proportions of total detected caesium. The chemical shifts of the two intracellular compartments were 2.44 ± 0.07 and 1.21 ± 0.18 ppm, relative to the 133Cs signal from the perfusate [347]. Similarly, in vivo 133Cs NMR measurements of caesium uptake into cells of Arabidopsis thaliana exposed to a caesium stress revealed that intracellular caesium was distributed in two kinds of compartment [348]. Sterically stabilised superparamagnetic iron oxide nanoparticles (SPIONs) were explored as potential MRI contrasting agents by 133Cs NMR. Thus, SPIONs were incubated with fresh human erythrocytes and the chemical shift and linewidth of 133Cs resonances from inside and outside the erythrocytes in 133Cs NMR spectra were monitored as a function of time. Results suggested that the SPIONS did not enter the erythrocytes and that their relaxation properties showed promise for use as MRI contrasting agents [349].
2.30. Platinum (195Pt)
There is one NMR-active isotope of platinum, spin-1/2 195Pt (natural abundance 33.83%), which has medium sensitivity and produces relatively narrow signals over a very wide chemical shift range (-6500 to 200 ppm). There are a few applications of 195Pt NMR with biological samples. In a study on the human metabolism of the anticancer drug iproplatin, a 195Pt NMR analysis of urine from patients receiving a high dose of iproplatin showed that platinum species existed as divalent complexes and not quadrivalent complexes [350]. 195Pt NMR has also been used for in vivo monitoring of the local disposition kinetics of the anticancer drug carboplatin after subcutaneous injection in rats. This used a surface coil of 2 cm diameter tuned to 18.3 MHz that was placed over the injection site (back of the neck of the animals) and experiments were optimised by in vitro 195Pt NMR measurements on model solutions of potassium tetrachloroplatinate(II), carboplatin, and cisplatin with different solvents including H2O, DMSO and DMF. In vivo 195Pt NMR spectra showed a broad resonance at -1715 ± 8 ppm and the elimination rate constant of local disposition of carboplatin was 0.017 min-1 [351].
2.31. Mercury (199Hg)
There are two NMR-active isotopes of mercury, spin-1/2 199Hg (natural abundance 16.87%) and spin-3/2 201Hg (natural abundance 13.18%), which have a very wide chemical shift range (-3000 to 500 ppm). 199Hg has low sensitivity, but produces sharp signals and is preferred over 201Hg, which produces very broad signals. There have been a few applications of 199Hg NMR with biological samples. Interactions of mercury chloride with membranes have been studied by 199Hg NMR. There was evidence for complexation with phosphatidylethanolamine (PE), phosphatidylserine (PS) and human erythrocyte membranes, whilst Hg(II) did not form complexes with egg phosphatidylcholine (PC) membranes. Interaction with PE and PS model membranes was described by the presence of two mercury sites, one labile and the other unlabile, on the NMR time scale. Calculated thermodynamic data indicated that PE is a better complexing agent than PS and mercury complexation with ligands or membranes was completely reversed by addition of decimolar NaCl solutions [352]. 199Hg NMR was used to obtain structural information about the metal receptor site of the MerR metalloregulatory protein alone and in a complex with the regulatory target, DNA. One- and two-dimensional NMR data were consistent with a trigonal planar mercury-thiolate co-ordination environment consisting only of cysteine side chains and they resolved structural details about metal ion recognition and the allosteric mechanism [353]. 199Hg NMR was used to confirm that in the structure of the mercury-bound form of the bacterial mercury detoxification system MerP, Hg(II) is bicoordinate with cysteine side chains [354].
2.32. Thallium (205Tl)
There are two NMR-active isotopes of thallium, 203Tl (natural abundance 29.52%) and 205Tl (natural abundance 70.48%), which are both spin-1/2. 205Tl is the preferred nucleus and there have been a number of applications with biological systems and samples. 205Tl NMR was used to monitor the binding of thallium ions to gramicidins A, B and C in aqueous dispersions of lysophosphatidylcholine. For 5 mM gramicidin dimer in the presence of 100 mM lysophosphatidylcholine, only 50% or less of the gramicidin appeared to be accessible to thallium. Analysis of the 205Tl chemical shift as a function of thallium ion concentration over the range 0.65-50 mM indicated that only one thallium ion can be bound by the gramicidins under these conditions. The thallium equilibrium binding constants were 582 ± 20 M-1, 1949 ± 100 M-1 and 390 ± 20 M-1 for gramicidins A, B and C, respectively [355]. The interaction between thallous ions and gramicidin A in DMPC vesicles was also studied by 205Tl NMR. Results showed that only multiple-channel occupancy can account for the observed chemical shifts and the data were analysed to give the equilibrium association constants of 450-600 and 5-20 M-1 for the binding of the first and the second ions, respectively, at a temperature of 34 ºC [356]. 205Tl NMR analysis of enzyme-bound thallium was used to characterise the structure of the monovalent cation activator binding site of S-adenosylmethionine synthetase from Escherichia coli. The chemical shift of the enzyme-thallium complex is 176 ppm downfield from aqueous thallium. The 205Tl resonance shifts upfield to 85 ppm in the enzyme-Mg(II)-thallium complex, to 38 ppm in the enzyme-thallium-AdoMet complex and to 34 ppm in the enzyme-thallium-AdoMet-Mg(II) complex. The NMR data suggested that the substrates or products of the enzyme do not coordinate to the monovalent cation activator, implying that monovalent cation activation results from alterations in protein conformation [357]. 205Tl NMR was used to probe the monovalent cation binding sites of bovine plasma activated protein C and des-1-41-light-chain-activated protein C [358]. 205Tl NMR has been used to study the binding of thallium ions to human serotransferrin and chicken ovotransferrin in the presence of carbonate and oxalate. With carbonate as the synergistic anion, two 205Tl NMR signals were observed that originate from the bound metal ion in the two high-affinity iron-binding sites of each protein. Titration experiments showed that thallium ions are bound with a greater affinity at the C-terminal site of serotransferrin, whilst there was no site preference for ovotransferrin. When oxalate was used as the anion instead of carbonate, the 205Tl NMR signals originating from the bound metal ion in the sites of ovotransferrin were shifted downfield and became almost degenerate [359]. Conformational changes in wild-type and mutant forms of yeast pyruvate kinase have been investigated by 205Tl NMR [360,361]. 205Tl NMR methods have also been developed for the characterisation of monovalent cation binding to nucleic acids, including the first 1H-205Tl scalar couplings observed in a biological system [362]. 205Tl solid-state NMR has been used to characterise cation binding in Na+,K+-ATPase using thallium ions as a substitute for potassium ions. 205Tl signals originating from occluded thallium and from non-specifically bound thallium were detected and distinguished (Figure 27).
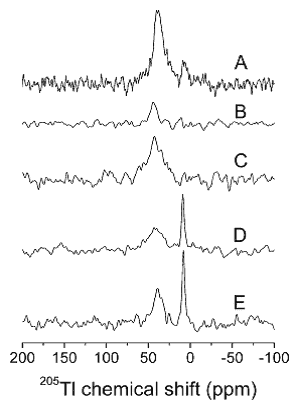
Figure 27. 205Tl MAS NMR spectra of Tl+ interacting with Na+, K±ATPase from shark rectal glands. In all five samples, the protein concentration was about 30 mg/mL in the rotor. For experiment A, the sample contains about 9 mol Tl+ per mol enzyme, and the integrated intensity corresponds to about 7 mol Tl+ per mol enzyme. In experiments B-E, the sample contains 3.3 mol Tl+ per mol enzyme. For experiment B, the integrated intensity corresponds to about 0.8 mol Tl+ per mol enzyme. Experiment C is identical to B, except that 1H decoupling was employed during data collection, and the integrated intensity was 3.2 mol Tl+ per mol enzyme. Experiments D and E are similar to B and C but with the addition of a 100-fold excess of K+ (330 mol K+ per mol enzyme). The integrated signal intensities correspond to 2.9 (D) and 3.4 mol Tl+ per mol enzyme (E). The experiments were performed at 17 °C at pH 6.9 in a 20 mM histidine and 1.3 mM CDTA buffer. Spectra B-E have the same vertical scale, and spectrum A has been reduced 2.2-fold in order to ease comparison. This Figure was reproduced with permission from Jakobsen et al. 2006 [363]; copyright © 2006 by American Chemical Society.
The effects of dipole-dipole coupling between 1H and 205Tl in the occlusion sites showed that the thallium ions are rigidly bound, rather than just occluded. A low chemical shift also suggested that occlusion site geometries have a relatively small contribution from carboxylate and hydroxyl groups. The non-specific binding of thallium ions were characterised by rapid chemical exchange, in agreement with an observed low binding affinity [363].
2.33. Lead (207Pb)
Lead has one NMR-active isotope, the spin-1/2 207Pb (natural abundance 22.1%), which has medium sensitivity and produces narrow signals over an extremely wide chemical shift range (-5500 to 6000 ppm). There appears to be only one published application of 207Pb NMR with a biological sample. 207Pb NMR has revealed that Pb(II) coordinates with the thiol-rich biomolecules glutathione and tris cysteine zinc finger proteins with trigonal pyramidal geometry (PbS3). Thus, glutathione binds Pb(II) with PbS3 geometry at pH 7.5 or higher with a 1:3 molar ratio of Pb(II) to glutathione. The zinc binding domain from HIV nucleocapsid protein (HIV-CCHC) appeared to bind Pb(II) in two PbS3 structures (5790 and 5744 ppm), one of which was more stable at high temperatures. This observation was consistent with previous studies suggesting that the HIV-CCHH peptide does not fold properly to afford a PbS2N motif, because histidine does not bind to Pb(II) [364].
3. Conclusions
This article has provided an overview of the properties of 39 NMR active nuclei and their applications in NMR measurements with biological and biomedical systems and samples. The measurements include solution- and solid-state NMR, magnetic resonance spectroscopy and magnetic resonance imaging. Examples have been provided for the wide range of analyses and types of information that can be obtained by NMR measurements using different NMR active nuclei. The current and ongoing technological developments in all types of NMR measurements will continue to push the boundaries for achieving higher resolution and higher sensitivity chemical, structural, dynamic and imaging information, thus expanding the types and increasing the complexities of biological and biomedical systems and samples amenable to NMR investigation. This will provide information for metabolic profiling in greater detail, a better understanding of the structures, functions and molecular mechanisms of biomolecules, the design and pharmacokinetics of new drugs, and the detection and monitoring of human diseases and their therapies.
Conflict of interest
The author confirms that this article content has no conflict of interest.
References
Key Reference: 1, 2, 7, 48, 68, 131, 214, 231, 334, 336
- Levitt MH. Spin dynamics: Basics of nuclear magnetic resonance (2nd Edition). John Wiley & Sons, 2013.Reference Source
- Kalverda AP, Gowdy J, Thompson GS, Homans SW, Henderson PJ, Patching SG. TROSY NMR with a 52 kDa sugar transport protein and the binding of a small-molecule inhibitor. Mol Membr Biol. 2014;31(4): 131-140.PubMed
- Hiruma-Shimizu K, Kalverda AP, Henderson PJ, Homans SW, Patching SG. Synthesis of uniformly deuterated n-dodecyl-β-D-maltoside (d39-DDM) for solubilization of membrane proteins in TROSY NMR experiments. J Labelled Comp Radiopharm. 2014;57(14): 737-743.PubMed
- Hiruma-Shimizu K, Shimizu H, Thompson GS, Kalverda AP, Patching SG. Deuterated detergents for structural and functional studies of membrane proteins: Properties, chemical synthesis and applications. Mol Membr Biol. 2016. [Epub ahead of print].CrossRef PubMed
- Nand D, Cukkemane A, Becker S, Baldus M. Fractional deuteration applied to biomolecular solid-state NMR spectroscopy. J Biomol NMR. 2012;52(2): 91-101.CrossRef PubMed
- Chevelkov V, Rehbein K, Diehl A, Reif B. Ultrahigh resolution in proton solid-state NMR spectroscopy at high levels of deuteration. Angew Chem Int Ed Engl. 2006;45(23): 3878-3881.CrossRef PubMed
- Reif B. Ultra-high resolution in MAS solid-state NMR of perdeuterated proteins: implications for structure and dynamics. J Magn Reson. 2012;216: 1-12.CrossRef PubMed
- Mote KR, Madhu PK. Proton-detected solid-state NMR spectroscopy of fully protonated proteins at slow to moderate magic-angle spinning frequencies. J Magn Reson. 2015;261: 149-156.CrossRef PubMed
- Chevelkov V, Xiang S, Giller K, Becker S, Lange A, Reif B. Perspectives for sensitivity enhancement in proton-detected solid-state NMR of highly deuterated proteins by preserving water magnetization. J Biomol NMR. 2015;61(2): 151-160.CrossRef PubMed
- Chevelkov V, Habenstein B, Loquet A, Giller K, Becker S, Lange A. Proton-detected MAS NMR experiments based on dipolar transfers for backbone assignment of highly deuterated proteins. J Magn Reson. 2014;242: 180-188.CrossRef PubMed
- Xiang S, Chevelkov V, Becker S, Lange A. Towards automatic protein backbone assignment using proton-detected 4D solid-state NMR data. J Biomol NMR. 2014;60(2-3): 85-90.CrossRef PubMed
- Deschamps M. Ultrafast magic angle spinning nuclear magnetic resonance. Ann Rep NMR Spectros. 2014;81: 109-144.CrossRef
- Zhou DH, Nieuwkoop AJ, Berthold DA, et al. Solid-state NMR analysis of membrane proteins and protein aggregates by proton detected spectroscopy. J Biomol NMR. 2012;54(3): 291-305.CrossRef PubMed
- Andreas LB, Le Marchand T, Jaudzems K, Pintacuda G. High-resolution proton-detected NMR of proteins at very fast MAS. J Magn Reson. 2015;253: 36-49.CrossRef PubMed
- Nieuwkoop AJ, Franks WT, Rehbein K, et al. Sensitivity and resolution of proton detected spectra of a deuterated protein at 40 and 60 kHz magic-angle-spinning. J Biomol NMR. 2015;61(2): 161-171.CrossRef PubMed
- Zhang R, Ramamoorthy A. Selective excitation enables assignment of proton resonances and 1H-1H distance measurement in ultrafast magic angle spinning solid state NMR spectroscopy. J Chem Phys. 2015;143(3): 034201.CrossRef PubMed
- Delgado-Goñi T, Campo S, Martín-Sitjar J, Cabañas ME, San Segundo B, Arús C. Assessment of a 1H high-resolution magic angle spinning NMR spectroscopy procedure for free sugars quantification in intact plant tissue. Planta. 2013;238(2): 397-413.PubMed
- Barbet-Massin E, Felletti M, Schneider R, et al. Insights into the structure and dynamics of measles virus nucleocapsids by 1H-detected solid-state NMR. Biophys J. 2014;107(4): 941-946.CrossRef PubMed
- Mroue KH, Nishiyama Y, Kumar Pandey M, et al. Proton-detected solid-state NMR spectroscopy of bone with ultrafast magic angle spinning. Sci Rep. 2015;5: 11991.CrossRef PubMed
- Singh C, Rai RK, Kayastha AM, Sinha N. Ultra fast magic angle spinning solid-state NMR spectroscopy of intact bone. Magn Reson Chem. 2016;54(2): 132-135.CrossRef PubMed
- Pandey MK, Zhang R, Hashi K, et al. 1020 MHz single-channel proton fast magic angle spinning solid-state NMR spectroscopy. J Magn Reson. 2015;261: 1-5.CrossRef PubMed
- Brauckmann JO, Janssen JW, Kentgens AP. High resolution triple resonance micro magic angle spinning NMR spectroscopy of nanoliter sample volumes. Phys Chem Chem Phys. 2016;18(6): 4902-4910.CrossRef PubMed
- Le Gall G. NMR spectroscopy of biofluids and extracts. Methods Mol Biol. 2015;1277: 29-36.CrossRef PubMed
- Dona AC, Jiménez B, Schäfer H, et al. Precision high-throughput proton NMR spectroscopy of human urine, serum, and plasma for large-scale metabolic phenotyping. Anal Chem. 2014;86(19): 9887-9894. CrossRef PubMed
- de Graaf RA, Prinsen H, Giannini C, Caprio S, Herzog RI. Quantification of 1H NMR spectra from human plasma. Metabolomics. 2015;11(6): 1702-1707.CrossRef PubMed
- Nadal-Desbarats L, Aïdoud N, Emond P, et al. Combined 1H-NMR and 1H-13C HSQC-NMR to improve urinary screening in autism spectrum disorders. Analyst. 2014;139(13): 3460-3468.CrossRef PubMed
- Lalande J, Halley H, Balayssac S, et al. 1H NMR metabolomic signatures in five brain regions of the AβPPswe Tg2576 mouse model of Alzheimer’s disease at four ages. J Alzheimers Dis. 2014;39(1): 121-143.PubMed
- Brugnara L, Mallol R, Ribalta J, et al. Improving assessment of lipoprotein profile in type 1 diabetes by 1H NMR spectroscopy. PLoS One. 2015;10(8): e0136348.CrossRef PubMed
- Ye N, Liu C, Shi P. Metabolomics analysis of cervical cancer, cervical intraepithelial neoplasia and chronic cervicitis by 1H NMR spectroscopy. Eur J Gynaecol Oncol. 2015;36(2): 174-180.PubMed
- Garcia-Simon M, Morales JM, Modesto-Alapont V, et al. Prognosis biomarkers of severe sepsis and septic shock by 1H NMR urine metabolomics in the Intensive Care Unit. PLoS One. 2015;10(11): e0140993.CrossRef PubMed
- Kyriakides M, Rama N, Sidhu J, Gabra H, Keun HC, El-Bahrawy M. Metabonomic analysis of ovarian tumour cyst fluid by proton nuclear magnetic resonance spectroscopy. Oncotarget. 2016;7(6): 7216-7226.PubMed
- Chalkias A, Fanos V, Noto A, et al. 1H NMR-metabolomics: can they be a useful tool in our understanding of cardiac arrest? Resuscitation. 2014;85(5): 595-601.CrossRef PubMed
- Rankin NJ, Preiss D, Welsh P, et al. The emergence of proton nuclear magnetic resonance metabolomics in the cardiovascular arena as viewed from a clinical perspective. Atherosclerosis. 2014;237(1): 287-300.CrossRef PubMed
- Sogin EM, Anderson P, Williams P, Chen CS, Gates RD. Application of 1H-NMR metabolomic profiling for reef-building corals. PLoS One. 2014;9(10): e111274.CrossRef PubMed
- Scano P, Rosa A, Incani A, et al. 1H NMR brain metabonomics of scrapie exposed sheep. Mol Biosyst. 2015;11(7): 2008-2016.CrossRef PubMed
- Chauton MS, Galloway TF, Kjørsvik E, et al. 1H NMR metabolic profiling of cod (Gadus morhua) larvae: potential effects of temperature and diet composition during early developmental stages. Biol Open. 2015;4(12): 1671-1678.CrossRef PubMed
- Beauclercq S, Nadal-Desbarats L, Hennequet-Antier C, et al. Serum and muscle metabolomics for the prediction of ultimate pH, a key factor for chicken-meat quality. J Proteome Res. 2016 15(4): 1168-1178.PubMed
- Zhao L, Huang Y, Hu J, Zhou H, Adeleye AS, Keller AA. 1H NMR and GC-MS based metabolomics reveal defense and detoxification mechanism of cucumber plant under nano-Cu stress. Environ Sci Technol. 2016;50(4): 2000-2010.CrossRef PubMed
- Brown MF, Lope-Piedrafita S, Martinez GV, Petrache HI. Solid-state deuterium NMR spectroscopy of membranes. In Webb GA (Ed) Modern magnetic resonance. Springer, Netherlands, 2006: 245-256.Reference Source
- Davis JH. Membranes: Deuterium NMR. eMagRes. 2007.CrossRef
- Yasuda T, Tsuchikawa H, Murata M, Matsumori N. Deuterium NMR of raft model membranes reveals domain-specific order profiles and compositional distribution. Biophys J. 2015;108(10): 2502-2506.CrossRef PubMed
- Kinnun JJ, Mallikarjunaiah KJ, Petrache HI, Brown MF. Elastic deformation and area per lipid of membranes: atomistic view from solid-state deuterium NMR spectroscopy. Biochim Biophys Acta. 2015;1848(1 Pt B): 246-259.CrossRef PubMed
- Tian X, Pavlopoulos S, Yang DP, Makriyannis A. The interaction of cannabinoid receptor agonists, CP55940 and WIN55212-2 with membranes using solid state 2H NMR. Biochim Biophys Acta. 2011;1808(9): 2095-2101.CrossRef PubMed
- Salnikov ES, Mason AJ, Bechinger B. Membrane order perturbation in the presence of antimicrobial peptides by 2H solid-state NMR spectroscopy. Biochimie. 2009;91(6): 734-743.CrossRef PubMed
- Kwon B, Waring AJ, Hong M. A 2H solid-state NMR study of lipid clustering by cationic antimicrobial and cell-penetrating peptides in model bacterial membranes. Biophys J. 2013;105(10): 2333-2342. CrossRef PubMed
- Pius J, Morrow MR, Booth V. ²H solid-state nuclear magnetic resonance investigation of whole Escherichia coli interacting with antimicrobial peptide MSI-78. Biochemistry. 2012;51(1): 118-125. PubMed
- Strandberg E, Esteban-Martín S, Salgado J, Ulrich AS. Orientation and dynamics of peptides in membranes calculated from 2H-NMR data. Biophys J. 2009;96(8): 3223-3232.CrossRef PubMed
- Cady SD, Schmidt-Rohr K, Wang J, Soto CS, Degrado WF, Hong M. Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 2010;463(7281): 689-692. CrossRef PubMed
- Wright AK, Batsomboon P, Dai J, Hung I, Zhou HX, Dudley GB, Cross TA. Differential binding of rimantadine enantiomers to influenza A M2 proton channel. J Am Chem Soc. 2016;138(5): 1506-1509.CrossRef PubMed
- Abu-Baker S, Lu JX, Chu S, Brinn CC, Makaroff CA, Lorigan GA. Side chain and backbone dynamics of phospholamban in phospholipid bilayers utilizing 2H and 15N solid-state NMR spectroscopy. Biochemistry. 2007;46(42): 11695-11706.PubMed
- Struts AV, Chawla U, Perera SM, Brown MF. Investigation of rhodopsin dynamics in its signaling state by solid-state deuterium NMR spectroscopy. Methods Mol Biol. 2015;1271: 133-158. CrossRef PubMed
- Shi X, Holland GP, Yarger JL. Molecular dynamics of spider dragline silk fiber investigated by 2H MAS NMR. Biomacromolecules. 2015;16(3): 852-859.PubMed
- Lalli D, Schanda P, Chowdhury A, et al. Three-dimensional deuterium-carbon correlation experiments for high-resolution solid-state MAS NMR spectroscopy of large proteins. J Biomol NMR. 2011;51(4): 477-485.CrossRef PubMed
- Kaieda S, Setlow B, Setlow P, Halle B. Mobility of core water in Bacillus subtilis spores by 2H NMR. Biophys J. 2013;105(9): 2016-2023.CrossRef PubMed
- Friedline AW, Zachariah MM, Johnson K, et al. Water behavior in bacterial spores by deuterium NMR spectroscopy. J Phys Chem B. 2014;118(30): 8945-8955.CrossRef PubMed
- Yuen AK, Lafon O, Charpentier T, et al. Measurement of long-range interatomic distances by solid-state tritium-NMR spectroscopy. J Am Chem Soc. 2010;132(6): 1734-1735.CrossRef PubMed
- Newmark RD, Un S, Williams PG, Carson PJ, Morimoto H, Klein MP. 3H nuclear magnetic resonance study of anaerobic glycolysis in packed erythrocytes. Proc Natl Acad Sci U S A. 1990;87(2): 583-587.CrossRef PubMed
- Dive V, Lai A, Valensin G, Saba G, Yiotakis A, Toma F. Proton and tritium NMR relaxation studies of peptide inhibitor binding to bacterial collagenase: conformation and dynamics. Biopolymers. 1991;15;31(3): 305-317.CrossRef PubMed
- Gehring K, Williams PG, Pelton JG, Morimoto H, Wemmer DE. Tritium NMR spectroscopy of ligand binding to maltose-binding protein. Biochemistry. 1991;30(22): 5524-5531.CrossRef PubMed
- Kubinec MG, Culf AS, Cho H, et al. Applications of tritium NMR to macromolecules: a study of two nucleic acid molecules. J Biomol NMR. 1996;7(3): 236-246.CrossRef PubMed
- Culf AS, Gerig JT, Williams PG. Tritium NMR studies of the human carbonic anhydrase I-benzenesulfonamide complex. J Biomol NMR. 1997;10(3): 293-299.PubMed
- Jackowski K, Jaszuński M, Kamieński B, Wilczek M. NMR frequency and magnetic dipole moment of 3He nucleus. J Magn Reson. 2008;193(1): 147-149.CrossRef PubMed
- Leawoods JC, Yablonskiy DA, Saam B, Gierada DS, Conradi MS. (2001). Hyperpolarized 3He gas production and MR imaging of the lung. Concepts Magn Reson. 2001;13: 277-293.Reference Source
- Bidinosti CP, Choukeife J, Nacher PJ, Tastevin G. In vivo NMR of hyperpolarized 3He in the human lung at very low magnetic fields. J Magn Reson. 2003;162(1): 122-132.CrossRef PubMed
- Lipson DA, van Beek EJR. Noble gas ventilation imaging using 3He and 129Xe. In Lipson DA, van Beek EJR (Eds) Functional Lung Imaging. Taylor & Francis, 2005.Reference Source
- Fain SB, Korosec FR, Holmes JH, O’Halloran R, Sorkness RL, Grist TM. Functional lung imaging using hyperpolarized gas MRI. J Magn Reson Imaging. 2007;25: 910-923.CrossRef PubMed
- Kauczor HU. MRI of the lung. In Baert AL, Knauth M, Sartor K (Eds) Medical radiology: Diagnostic imaging. Springer, 2009.Reference Source
- Fain S, Schiebler ML, McCormack DG, Parraga G. Imaging of lung function using hyperpolarized helium-3 magnetic resonance imaging: Review of current and emerging translational methods and applications. J Magn Reson Imaging. 2010;32(6): 1398-1408. CrossRef PubMed
- Komoroski RA. Applications of 7Li NMR in biomedicine. Magn Reson Imaging. 2000;18(2): 103-116.CrossRef PubMed
- Komoroski RA. Biomedical applications of 7Li NMR. NMR Biomed. 2005;18(2): 67-73.PubMed
- Ramaprasad S, Ripp E, Pi J, Lyon M. Pharmacokinetics of lithium in rat brain regions by spectroscopic imaging. Magn Reson Imaging. 2005;23(8): 859-863.CrossRef PubMed
- Forester BP, Finn CT, Berlow YA, Wardrop M, Renshaw PF, Moore CM. Brain lithium, N-acetyl aspartate and myo-inositol levels in older adults with bipolar disorder treated with lithium: a lithium-7 and proton magnetic resonance spectroscopy study. Bipolar Disord. 2008;10(6): 691-700.PubMed
- Smith FE, Cousins DA, Thelwall PE, Ferrier IN, Blamire AM. Quantitative lithium magnetic resonance spectroscopy in the normal human brain on a 3 T clinical scanner. Magn Reson Med. 2011;66(4): 945-949.CrossRef PubMed
- Port JD, Rampton KE, Shu Y, Manduca A, Frye MA. Short TE 7Li-MRS confirms Bi-exponential lithium T2 relaxation in humans and clearly delineates two patient subtypes. J Magn Reson Imaging. 2013;37(6): 1451-1459.PubMed
- Komoroski RA, Lindquist DM, Pearce JM. Lithium compartmentation in brain by 7Li MRS: effect of total lithium concentration. NMR Biomed. 2013;26(9): 1152-1157.PubMed
- Machado-Vieira R, Otaduy MC, et al. A selective association between central and peripheral lithium levels in remitters in bipolar depression: A 3T-7Li magnetic resonance spectroscopy study. Acta Psychiatr Scand. 2016;133(3): 214-220.CrossRef PubMed
- Haimovich A, Eliav U, Goldbourt A. Determination of the lithium binding site in inositol monophosphatase, the putative target for lithium therapy, by magic-angle-spinning solid-state NMR. J Am Chem Soc. 2012;134(12): 5647-5651.CrossRef PubMed
- Király P. Background-free solution boron NMR spectroscopy. Magn Reson Chem. 2012;50(9): 620-626.PubMed
- Bendel P. Biomedical applications of 10B and 11B NMR. NMR Biomed. 2005;18(2): 74-82.PubMed
- Wittig A, Michel J, Moss RL, et al. Boron analysis and boron imaging in biological materials for Boron Neutron Capture Therapy (BNCT). Crit Rev Oncol Hematol. 2008;68(1): 66-90.Reference Source. Erratum in Crit Rev Oncol Hematol. 2008;68(1): 91.Reference Source
- Bonora M, Corti M, Borsa F, et al. 1H and 10B NMR and MRI investigation of boron- and gadolinium-boron compounds in boron neutron capture therapy. Appl Radiat Isot. 2011;69(12): 1702-1705.CrossRef PubMed
- Yoshino K, Yabe T, Hattori T, Saito K, Ishikawa A, Ohki H. 10B-NMR determination of 10B-BPA, 10B-BPA-fructose complex and total 10B in blood for BNCT. Appl Radiat Isot. 2014;88: 74-77.CrossRef PubMed
- Cusack M, Kamenos NA, Rollion-Bard C, Tricot G. Red coralline algae assessed as marine pH proxies using 11B MAS NMR. Sci Rep. 2015;5: 8175.CrossRef PubMed
- Jaipuria G, Lobo NP, Shet D, Atreya HS. High resolution methyl selective 13C-NMR of proteins in solution and solid state. J Biomol NMR. 2012;54(1): 33-42.PubMed
- Felli IC, Pierattelli R. Spin-state-selective methods in solution- and solid-state biomolecular 13C NMR. Prog Nucl Magn Reson Spectrosc. 2015;84-85: 1-13.CrossRef PubMed
- Bertini I, Felli IC, Gonnelli L, Kumar M V V, Pierattelli R. 13C direct-detection biomolecular NMR spectroscopy in living cells. Angew Chem Int Ed Engl. 2011;50(10): 2339-2341.CrossRef PubMed
- de Graaf RA, Rothman DL, Behar KL. State of the art direct 13C and indirect 1H-[13C] NMR spectroscopy in vivo. A practical guide. NMR Biomed. 2011;24(8): 958-972.CrossRef PubMed
- Rodrigues TB, Valette J, Bouzier-Sore AK. 13C NMR spectroscopy applications to brain energy metabolism. Front Neuroenergetics. 2013;5: 9.CrossRef PubMed
- Xin L, Lanz B, Lei H, Gruetter R. Assessment of metabolic fluxes in the mouse brain in vivo using 1H-[13C] NMR spectroscopy at 14.1 Tesla. J Cereb Blood Flow Metab. 2015;35(5): 759-765.CrossRef PubMed
- Marin-Valencia I, Hooshyar MA, Pichumani K, Sherry AD, Malloy CR. The ratio of acetate-to-glucose oxidation in astrocytes from a single 13C NMR spectrum of cerebral cortex. J Neurochem. 2015;132(1): 99-109.PubMed
- Shen J. Modeling the glutamate-glutamine neurotransmitter cycle. Front Neuroenergetics. 2013;5: 1.Reference Source
- Ramadan S, Lin A, Stanwell P. Glutamate and glutamine: a review of in vivo MRS in the human brain. NMR Biomed. 2013;26(12): 1630-1646.PubMed
- Patching SG. Roles of facilitative glucose transporter GLUT1 in [18F]FDG positron emission tomography (PET) imaging of human diseases. J Diagn Imag Ther. 2015;2: 30-102.CrossRef
- Jalloh I, Carpenter KL, Helmy A, Carpenter TA, Menon DK, Hutchinson PJ. Glucose metabolism following human traumatic brain injury: methods of assessment and pathophysiological findings. Metab Brain Dis. 2015;30(3): 615-632.CrossRef PubMed
- Gallagher CN, Carpenter KL, Grice P, et al. The human brain utilizes lactate via the tricarboxylic acid cycle: a 13C-labelled microdialysis and high-resolution nuclear magnetic resonance study. Brain. 2009;132(10): 2839-2849.CrossRef PubMed
- Carpenter KL, Jalloh I, Gallagher CN, et al. 13C-labelled microdialysis studies of cerebral metabolism in TBI patients. Eur J Pharm Sci. 2014;57: 87-97.CrossRef PubMed
- Zheng H, Zheng Y, Wang D, et al. Analysis of neuron-astrocyte metabolic cooperation in the brain of db/db mice with cognitive decline using 13C NMR spectroscopy. J Cereb Blood Flow Metab. 2016. [Epub ahead of print]CrossRef PubMed
- Patching SG. Solid-state NMR structures of integral membrane proteins. Mol Membr Biol. 2016. [Epub ahead of print]CrossRef PubMed
- Raleigh DP, Levitt MH, Griffin RG. Rotational resonance in solid state NMR. Chem Phys Lett. 1988;146(1-2): 71-76.CrossRef
- Gullion T, Schaefer J. Rotational-echo double-resonance NMR. J Magn Reson. 1989;81: 196.CrossRef
- Patching SG, Brough AR, Herbert RB, Rajakarier JA, Henderson PJ, Middleton DA. Substrate affinities for membrane transport proteins determined by 13C cross-polarization magic-angle spinning nuclear magnetic resonance spectroscopy. J Am Chem Soc. 2004;126: 3072-3080.PubMed
- Patching SG, Herbert RB, O’Reilly J, Brough AR, Henderson PJ. Low 13C-background for NMR-based studies of ligand binding using 13C-depleted glucose as carbon source for microbial growth: 13C-labeled glucose and 13C-forskolin binding to the galactose-H+ symport protein GalP in Escherichia coli. J Am Chem Soc. 2004;126: 86-87.PubMed
- Patching SG, Psakis G, Baldwin SA, Baldwin J, Henderson PJ, Middleton DA. Relative substrate affinities of wild-type and mutant forms of the Escherichia coli sugar transporter GalP determined by solid-state NMR. Mol Membr Biol. 2008;25: 474-484.PubMed
- Patching SG, Henderson PJ, Sharples DJ, Middleton DA. Probing the contacts of a low-affinity substrate with a membrane-embedded transport protein using 1H-13C cross-polarisation magic-angle spinning solid-state NMR. Mol Membr Biol. 2013;30: 129-137. PubMed
- Williamson PT, Verhoeven A, Miller KW, Meier BH, Watts A. The conformation of acetylcholine at its target site in the membrane embedded nicotinic acetylcholine receptor. Proc Natl Acad Sci USA. 2007;104: 18031-18036.CrossRef PubMed
- Lopez JJ, Shukla AK, Reinhart C, Schwalbe H, Michel H, Glaubitz C. The structure of the neuropeptide bradykinin bound to the human G-protein coupled receptor bradykinin B2 as determined by solid-state NMR spectroscopy. Angew Chem Int Ed. 2008;47: 1668-1671. CrossRef PubMed
- Patching SG, Henderson PJ, Herbert RB, Middleton DA. Solid-state NMR spectroscopy detects interactions between tryptophan residues of the E. coli sugar transporter GalP and the alpha-anomer of the D-glucose substrate. J Am Chem Soc. 2008;130: 1236-1244.PubMed
- Edwards R, Madine J, Fielding L, Middleton DA. Measurement of multiple torsional angles from one-dimensional solid-state NMR spectra: application to the conformational analysis of a ligand in its biological receptor site. Phys Chem Chem Phys. 2010;12: 13999-14008.CrossRef PubMed
- Middleton DA, Hughes E, Esmann M. The conformation of ATP within the Na, K-ATPase nucleotide site: a statistically constrained analysis of REDOR solid-state NMR data. Angew Chem Int Ed. 2011;50: 7041-7044.CrossRef PubMed
- Lakatos A, Mörs K, Glaubitz C. How to investigate interactions between membrane proteins and ligands by solid-state NMR. Methods Mol Biol. 2012;914: 65-86.CrossRef PubMed
- Whittaker CA, Patching SG, Esmann M, Middleton DA. Ligand orientation in a membrane-embedded receptor site revealed by solid-state NMR with paramagnetic relaxation enhancement. Org Biomol Chem. 2015;13: 2664-2668.CrossRef PubMed
- Watts A. Solid-state NMR in drug design and discovery for membrane-embedded targets. Nat Rev Drug Discov. 2005;4: 555-568.CrossRef PubMed
- Williamson PTF. Solid-state NMR for the analysis of high-affinity ligand/receptor interactions. Concept Magn Reson A. 2009;34A: 144-172.CrossRef
- Tapaneeyakorn S, Goddard AD, Oates J, Willis CL, Watts A. Solution- and solid-state NMR studies of GPCRs and their ligands. Biochim Biophys Acta. 2011;1808: 1462-1475.CrossRef PubMed
- Ding X, Zhao X, Watts A. G-protein-coupled receptor structure, ligand binding and activation as studied by solid-state NMR spectroscopy. Biochem J. 2013;450: 443-457.PubMed
- Middleton DA, Patching SG. Solid-state NMR spectroscopy in drug design and discovery (Chapter 51). In Andrushko V, Andrushko N (Eds) Stereoselective synthesis of drugs and natural products. John Wiley & Sons Ltd, Chichester, UK, 2013.Reference Source
- Petkova AT, Tycko R. Rotational resonance in uniformly 13C-labeled solids: effects on high-resolution magic-angle spinning NMR spectra and applications in structural studies of biomolecular systems. J Magn Reson. 2004;168(1): 137-146.CrossRef PubMed
- Chou FC, Tsai TW, Cheng HM, Chan JC. Measurements of 13C multiple-quantum coherences in amyloid fibrils under magic-angle spinning. J Phys Chem B. 2012;116(24): 7162-7167.PubMed
- Itoh-Watanabe H, Kamihira-Ishijima M, Javkhlantugs N, et al. Role of aromatic residues in amyloid fibril formation of human calcitonin by solid-state 13C NMR and molecular dynamics simulation. Phys Chem Chem Phys. 2013;15(23): 8890-901.CrossRef PubMed
- Silverstein MC, Bilici K, Morgan SW, Wang Y, Zhang Y, Boutis GS. 13C, 2H NMR studies of structural and dynamical modifications of glucose-exposed porcine aortic elastin. Biophys J. 2015;108(7): 1758-1772.CrossRef PubMed
- Weber F, Böhme J, Scheidt HA, et al. 31P and 13C solid-state NMR spectroscopy to study collagen synthesis and biomineralization in polymer-based bone implants. NMR Biomed. 2012;25(3): 464-475.CrossRef PubMed
- Dick-Perez M, Wang T, Salazar A, Zabotina OA, Hong M. Multidimensional solid-state NMR studies of the structure and dynamics of pectic polysaccharides in uniformly 13C-labeled Arabidopsis primary cell walls. Magn Reson Chem. 2012;50(8): 539-550.CrossRef PubMed
- Wang T, Hong M. Solid-state NMR investigations of cellulose structure and interactions with matrix polysaccharides in plant primary cell walls. J Exp Bot. 2016;67(2): 503-514.CrossRef PubMed
- Popescu CM, Larsson PT, Tibirna CM, Vasile C. Characterization of fungal-degraded lime wood by X-ray diffraction and cross-polarization magic-angle-spinning 13C-nuclear magnetic resonance spectroscopy. Appl Spectrosc. 2010;64(9): 1054-1060.PubMed
- Peter-Valence F, Llarena-Hernandez C, Largeteau M, et al. Chemical characterization of the biomass of an edible medicinal mushroom, Agaricus subrufescens, via solid-state 13C NMR. J Agric Food Chem. 2011;59(16): 8939-8943.PubMed
- Kameda T, Tamada Y. Variable-temperature 13C solid-state NMR study of the molecular structure of honeybee wax and silk. Int J Biol Macromol. 2009;44(1): 64-69.CrossRef PubMed
- Forte C, Piazzi A, Pizzanelli S, Certini G. CP MAS 13C spectral editing and relative quantitation of a soil sample. Solid State Nucl Magn Reson. 2006;30(2): 81-88.CrossRef PubMed.
- Duarte RM, Fernández-Getino AP, Duarte AC. Humic acids as proxies for assessing different Mediterranean forest soils signatures using solid-state CPMAS 13C NMR spectroscopy. Chemosphere. 2013;91(11): 1556-1565.CrossRef PubMed
- Jiménez-González MA, De la Rosa JM, Jiménez-Morillo NT, Almendros G, González-Pérez JA, Knicker H. Post-fire recovery of soil organic matter in a Cambisol from typical Mediterranean forest in Southwestern Spain. Sci Total Environ. 2016. [Epub ahead of print]PubMed
- Takeuchi K, Arthanari H, Shimada I, Wagner G. Nitrogen detected TROSY at high field yields high resolution and sensitivity for protein NMR. J Biomol NMR. 2015;63(4): 323-331.CrossRef PubMed
- Takeuchi K, Arthanari H, Imai M, Wagner G, Shimada I. Nitrogen-detected TROSY yields comparable sensitivity to proton-detected TROSY for non-deuterated, large proteins under physiological salt conditions. J Biomol NMR. 2016;64(2): 143-151.CrossRef PubMed
- Hanashima S, Fujiwara N, Matsumoto K, et al. A solution 17O-NMR approach for observing an oxidized cysteine residue in Cu, Zn-superoxide dismutase. Chem Commun (Camb). 2013;49(14): 1449-1451.CrossRef PubMed
- Zhu XH, Zhang N, Zhang Y, Zhang X, Ugurbil K, Chen W. In vivo 17O NMR approaches f or brain study at high field. NMR Biomed. 2005;18(2): 83-103.PubMed
- Zhu XH, Zhang Y, Wiesner HM, Ugurbil K, Chen W. In vivo measurement of CBF using 17O NMR signal of metabolically produced H217O as a perfusion tracer. Magn Reson Med. 2013;70(2): 309-314.PubMed
- Fiat D, Hankiewicz J, Liu S, Trbovic S, Brint S. 17O magnetic resonance imaging of the human brain. Neurol Res. 2004;26(8): 803-808.CrossRef PubMed
- McCommis KS, He X, Abendschein DR, Gupte PM, Gropler RJ, Zheng J. Cardiac 17O MRI: toward direct quantification of myocardial oxygen consumption. Magn Reson Med. 2010;63(6): 1442-1447.CrossRef PubMed
- Narazaki M, Kanazawa Y, Koike S, Ando K, Ikehira H. Quantitative 17O imaging towards oxygen consumption study in tumor bearing mice at 7 T. Magn Reson Imaging. 2013;31(5): 643-650.CrossRef PubMed
- Borowiak R, Groebner J, Haas M, Hennig J, Bock M. Direct cerebral and cardiac 17O-MRI at 3 Tesla: initial results at natural abundance. MAGMA. 2014;27(1): 95-99.CrossRef PubMed
- Lemaître V, de Planque MR, Howes AP, Smith ME, Dupree R, Watts A. Solid-state 17O NMR as a probe for structural studies of proteins in biomembranes. J Am Chem Soc. 2004;126(47): 15320-15321.PubMed
- Wong A, Howes AP, Pike KJ, et al. New limits for solid-state 17O NMR spectroscopy: complete resolution of multiple oxygen sites in a simple biomolecule. J Am Chem Soc. 2006;128(24): 7744-7745.PubMed
- Wong A, Howes AP, Yates JR, et al. Ultra-high resolution 17O solid-state NMR spectroscopy of biomolecules: a comprehensive spectral analysis of monosodium L-glutamate·monohydrate. Phys Chem Chem Phys. 2011;13(26): 12213-12224.CrossRef PubMed
- Wong A, Beevers AJ, Kukol A, Dupree R, Smith ME. Solid-state 17O NMR spectroscopy of a phospholemman transmembrane domain protein: implications for the limits of detecting dilute 17O sites in biomaterials. Solid State Nucl Magn Reson. 2008;33(4): 72-75. CrossRef PubMed
- Antzutkin ON, Iuga D, Filippov AV, Kelly RT, Becker-Baldus J, Brown SP, Dupree R. Hydrogen bonding in Alzheimer’s amyloid-β fibrils probed by 15N{17O} REAPDOR solid-state NMR spectroscopy. Angew Chem Int Ed Engl. 2012;51(41): 10289-10292.PubMed
- Chen H, Viel S, Ziarelli F, Peng L. 19F NMR: a valuable tool for studying biological events. Chem Soc Rev. 2013;42(20): 7971-7982. CrossRef PubMed
- Yu JX, Hallac RR, Chiguru S, Mason RP. New frontiers and developing applications in 19F NMR. Prog Nucl Magn Reson Spectrosc. 2013;70: 25-49.CrossRef PubMed
- Marsh EN, Suzuki Y. Using 19F NMR to probe biological interactions of proteins and peptides. ACS Chem Biol. 2014;9(6): 1242-1250.PubMed
- Zhu L, Yang J, Li H, Sun H, Liu J, Wang J. Conformational change study of dengue virus NS2B-NS3 protease using 19F NMR spectroscopy. Biochem Biophys Res Commun. 2015;461(4): 677-680.CrossRef PubMed
- Arntson KE, Pomerantz WC. Protein-observed fluorine NMR: A bioorthogonal approach for small molecule discovery. J Med Chem. 2015. [Epub ahead of print].CrossRef PubMed
- Gee CT, Koleski EJ, Pomerantz WC. Fragment screening and druggability assessment for the CBP/p300 KIX domain through protein-observed 19F NMR spectroscopy. Angew Chem Int Ed Engl. 2015;54(12): 3735-3739.PubMed
- Veronesi M, Giacomina F, Romeo E, et al. Fluorine nuclear magnetic resonance-based assay in living mammalian cells. Anal Biochem. 2016;495: 52-59.CrossRef PubMed
- Tooyama I, Yanagisawa D, Taguchi H, et al. Amyloid imaging using fluorine-19 magnetic resonance imaging (19F-MRI). Ageing Res Rev. 2016. [Epub ahead of print].CrossRef PubMed
- Amiri H, Srinivas M, Veltien A, van Uden MJ, de Vries IJ, Heerschap A. Cell tracking using 19F magnetic resonance imaging: technical aspects and challenges towards clinical applications. Eur Radiol. 2015;25(3): 726-735.CrossRef PubMed
- Gaudet JM, Ribot EJ, Chen Y, Gilbert KM, Foster PJ. Tracking the fate of stem cell implants with fluorine-19 MRI. PLoS One. 2015;10(3): e0118544.CrossRef PubMed
- Rose LC, Kadayakkara DK, Wang G, et al. Fluorine-19 labeling of stromal vascular fraction cells for clinical imaging applications. Stem Cells Transl Med. 2015;4(12): 1472-1481.CrossRef PubMed
- Bartusik D, Aebisher D. 19F applications in drug development and imaging – a review. Biomed Pharmacother. 2014;68(6): 813-817. CrossRef PubMed
- Hahn T, Kozerke S, Schwizer W, Fried M, Boesiger P, Steingoetter A. Real-time multipoint gastrointestinal 19-fluorine catheter tracking. Magn Reson Med. 2014;71(1): 302-307.CrossRef PubMed
- Wi S, Sinha N, Hong M. Long-range 1H-19F distance measurement in peptides by solid-state NMR. J Am Chem Soc. 2004;126(40): 12754-12755.PubMed
- Luo W, Mani R, Hong M. Side-chain conformation of the M2 transmembrane peptide proton channel of influenza a virus from 19F solid-state NMR. J Phys Chem B. 2007;111(36): 10825-10832. PubMed
- Witter R, Nozirov F, Sternberg U, Cross TA, Ulrich AS, Fu R. Solid-state 19F NMR spectroscopy reveals that Trp41 participates in the gating mechanism of the M2 proton channel of influenza A virus. J Am Chem Soc. 2008;130(3): 918-924.PubMed
- Grage SL, Xu X, Schmitt M, Wadhwani P, Ulrich AS. 19F-Labeling of peptides revealing long-range NMR distances in fluid membranes. J Phys Chem Lett. 2014;5(24): 4256-4259.CrossRef PubMed
- Huang W, Varani G, Drobny GP. 13C/15N-19F intermolecular REDOR NMR study of the interaction of TAR RNA with Tat peptides. J Am Chem Soc. 2010;132(50): 17643-17645.CrossRef PubMed
- Shi P, Li D, Chen H, Xiong Y, Wang Y, Tian C. In situ 19F NMR studies of an E. coli membrane protein. Protein Sci. 2012;21(4): 596-600.PubMed
- Wadhwani P, Strandberg E, Heidenreich N, Bürck J, Fanghänel S, Ulrich AS. Self-assembly of flexible β-strands into immobile amyloid-like β-sheets in membranes as revealed by solid-state 19F NMR. J Am Chem Soc. 2012;134(15): 6512-6515.PubMed
- Matsumori N, Kasai Y, Oishi T, Murata M, Nomura K. Orientation of fluorinated cholesterol in lipid bilayers analyzed by 19F tensor calculation and solid-state NMR. J Am Chem Soc. 2008;130(14): 4757-4766.PubMed
- Elgavish GA. Shift-reagent-aided 23Na NMR spectroscopy. Invest Radiol. 1989;24(12): 1028-1033.CrossRef PubMed
- Puckeridge M, Chapman BE, Conigrave AD, Kuchel PW. Quantitative model of NMR chemical shifts of 23Na+ induced by TmDOTP: applications in studies of Na+ transport in human erythrocytes. J Inorg Biochem. 2012;115: 211-219. Erratum in J Inorg Biochem. 2013;121: 196.CrossRef PubMed
- Ouwerkerk R, van Echteld CJ, Staal GE, Rijksen G. Erythrocyte Na+/K+ ATPase activity measured with 23Na NMR. Magn Reson Med. 1989;12(2): 164-171.PubMed
- Knubovets TL, Revazov AV, Sibeldina LA, Eichhoff U. 23Na NMR measurement of the maximal rate of active sodium efflux from human red blood cells. Magn Reson Med. 1989;9(2): 261-272.CrossRef PubMed
- Cacciafesta M, Marigliano V, Ferri C, et al. 23Na-NMR study of cation cotransport in human red blood cells. Am J Physiol. 1992;262(5 Pt 1): C1292-C1296.PubMed
- Martin JB, Klein G, Satre M. 23Na NMR study of intracellular sodium ions in Dictyostelium discoideum amoeba. Arch Biochem Biophys. 1987;254(2): 559-567.CrossRef PubMed
- Castle AM, Macnab RM, Shulman RG. Measurement of intracellular sodium concentration and sodium transport in Escherichia coli by 23Na nuclear magnetic resonance. J Biol Chem. 1986;261(7): 3288-3294.PubMed
- Górecki K, Hägerhäll C, Drakenberg T. The Na+ transport in gram-positive bacteria defect in the Mrp antiporter complex measured with 23Na nuclear magnetic resonance. Anal Biochem. 2014;445: 80-86.PubMed
- Shah NJ, Worthoff WA, Langen KJ. Imaging of sodium in the brain: a brief review. NMR Biomed. 2016;29(2): 162-174.CrossRef PubMed
- Bottomley PA. Sodium MRI in human heart: a review. NMR Biomed. 2016;29(2): 187-196.CrossRef PubMed
- Reetz K, Romanzetti S, Dogan I, et al. Increased brain tissue sodium concentration in Huntington’s Disease – a sodium imaging study at 4 T. Neuroimage. 2012;63(1): 517-524.PubMed
- Zaaraoui W, Konstandin S, Audoin B, et al. Distribution of brain sodium accumulation correlates with disability in multiple sclerosis: a cross-sectional 23Na MR imaging study. Radiology. 2012;264(3): 859-867.CrossRef PubMed
- Eisele P, Konstandin S, Griebe M, et al. Heterogeneity of acute multiple sclerosis lesions on sodium 23Na MRI. Mult Scler. 2015. [Epub ahead of print].CrossRef PubMed
- Haneder S, Ong MM, Budjan JM, et al. 23Na-magnetic resonance imaging of the human lumbar vertebral discs: in vivo measurements at 3.0 T in healthy volunteers and patients with low back pain. Spine J. 2014;14(7): 1343-1350.CrossRef PubMed
- Hammon M, Grossmann S, Linz P, et al. 23Na Magnetic resonance imaging of the lower leg of acute heart failure patients during diuretic treatment. PLoS One. 2015;10(10): e0141336.CrossRef PubMed
- Bock JL, Crull GB, Wishnia A, Springer CS Jr. 25Mg NMR studies of magnesium binding to erythrocyte constituents. J Inorg Biochem. 1991;44(2): 79-87.CrossRef PubMed
- Lee ME, Nowak T. 25Mg NMR studies of yeast enolase and rabbit muscle pyruvate kinase. Arch Biochem Biophys. 1992;293(2): 264-273.CrossRef PubMed
- Ehrlich RS, Colman RF. Cadmium-113 and magnesium-25 NMR study of the divalent metal binding sites of isocitrate dehydrogenases from pig heart. Biochim Biophys Acta. 1995;1246(2): 135-141. CrossRef PubMed
- Xian W, Tang JX, Janmey PA, Braunlin WH. The polyelectrolyte behavior of actin filaments: a 25Mg NMR study. Biochemistry. 1999;38(22): 7219-7226.CrossRef PubMed
- Kakitani Y, Koyama Y, Shimoikeda Y, et al. Stacking of bacteriochlorophyll c macrocycles in chlorosome from Chlorobium limicola as revealed by intermolecular 13C magnetic-dipole correlation, X-ray diffraction, and quadrupole coupling in 25Mg NMR. Biochemistry. 2009;48(1): 74-86.PubMed
- Lipton AS, Heck RW, Primak S, McNeill DR, Wilson DM 3rd, Ellis PD. Characterization of Mg2+ binding to the DNA repair protein apurinic/apyrimidic endonuclease 1 via solid-state 25Mg NMR spectroscopy. J Am Chem Soc. 2008;130(29): 9332-9341.PubMed
- Mazarguil H, Haran R, Laussac JP. The binding of aluminium to [Leu5]-enkephalin. An investigation using 1H, 13C and 27Al NMR spectroscopy. Biochim Biophys Acta. 1982;717(3): 465-472. CrossRef PubMed
- Kaspar A, Bilecen D, Scheffler K, Seelig J. Aluminum-27 nuclear magnetic resonance spectroscopy and imaging of the human gastric lumen. Magn Reson Med. 1996;36(2): 177-182.CrossRef PubMed
- Schwarz R, Kaspar A, Seelig J, Künnecke B. Gastrointestinal transit times in mice and humans measured with 27Al and 19F nuclear magnetic resonance. Magn Reson Med. 2002;48(2): 255-261. PubMed
- Rao KS, Easwaran KR. 27Al-NMR studies of aluminum transport across yeast cell membranes. Mol Cell Biochem. 1997;175(1-2): 59-63.PubMed
- Morita A, Yanagisawa O, Takatsu S, Maeda S, Hiradate S. Mechanism for the detoxification of aluminum in roots of tea plant (Camellia sinensis (L.) Kuntze). Phytochemistry. 2008;69(1): 147-153.CrossRef PubMed
- Morita A, Horie H, Fujii Y, Takatsu S, Watanabe N, Yagi A, Yokota H. Chemical forms of aluminum in xylem sap of tea plants (Camellia sinensis L.). Phytochemistry. 2004;65(20): 2775-2780. CrossRef PubMed
- Wang H, Chen RF, Iwashita T, Shen RF, Ma JF. Physiological characterization of aluminum tolerance and accumulation in tartary and wild buckwheat. New Phytol. 2015;205(1): 273-279.CrossRef PubMed
- Bryant PL, Lukiw WJ, Gan Z, Hall RW, Butler LG. High-field 19.6T 27Al solid-state MAS NMR of in vitro aluminated brain tissue. J Magn Reson. 2004;170(2): 257-262.CrossRef PubMed
- Bertermann R, Kröger N, Tacke R. Solid-state 29Si MAS NMR studies of diatoms: structural characterization of biosilica deposits. Anal Bioanal Chem. 2003;375(5): 630-634.PubMed
- Gröger C, Sumper M, Brunner E. Silicon uptake and metabolism of the marine diatom Thalassiosira pseudonana: Solid-state 29Si NMR and fluorescence microscopic studies. J Struct Biol. 2008;161(1): 55-63.PubMed
- La Vars SM, Johnston MR, Hayles J, et al. 29Si{1H} CP-MAS NMR comparison and ATR-FTIR spectroscopic analysis of the diatoms Chaetoceros muelleri and Thalassiosira pseudonana grown at different salinities. Anal Bioanal Chem. 2013;405(10): 3359-3365.CrossRef PubMed
- Prompers JJ, Wessels B, Kemp GJ, Nicolay K. MITOCHONDRIA: investigation of in vivo muscle mitochondrial function by 31P magnetic resonance spectroscopy. Int J Biochem Cell Biol. 2014;50: 67-72.CrossRef PubMed
- Kemp GJ, Ahmad RE, Nicolay K, Prompers JJ. Quantification of skeletal muscle mitochondrial function by 31P magnetic resonance spectroscopy techniques: a quantitative review. Acta Physiol (Oxf). 2015;213(1): 107-144.PubMed
- Phillips AC, Sleigh A, McAllister CJ, Brage S, Carpenter TA, Kemp GJ, Holland AJ. Defective mitochondrial function in vivo in skeletal muscle in adults with Down’s syndrome: a 31P-MRS study. PLoS One. 2013;8(12): e84031.CrossRef PubMed
- Schmidt TM, Wang ZJ, Keller S, et al. Postmortem 31P magnetic resonance spectroscopy of the skeletal muscle: α-ATP/Pi ratio as a forensic tool? Forensic Sci Int. 2014;242: 172-176.CrossRef PubMed
- Kamble RB, Peruvumba N J, Shivashankar R. Energy status and metabolism in intracranial space occupying lesions: a prospective 31P spectroscopic study. J Clin Diagn Res. 2014;8(11): RC05-RC08. CrossRef PubMed
- Plante DT, Trksak GH, Jensen JE, et al. Gray matter-specific changes in brain bioenergetics after acute sleep deprivation: a 31P magnetic resonance spectroscopy study at 4 Tesla. Sleep. 2014;37(12): 1919-1927. CrossRef PubMed
- Yuksel C, Tegin C, O’Connor L, et al. Phosphorus magnetic resonance spectroscopy studies in schizophrenia. J Psychiatr Res. 2015;68: 157-166.CrossRef PubMed
- Bubnovskaya L, Osinsky D, Trachevsky V, Naleskina L, Kovelskaya A, Gumenyuk L. Premorphological alterations in gastric mucosa in patients with gastric cancer: hypoxia level assessed by 31P NMR spectroscopy. Exp Oncol. 2014;36(4): 271-275.PubMed
- Zhang CY, Zhang Q, Zhang HM, Yang HS. 3.0T 31P MR spectroscopy in assessment of response to antiviral therapy for chronic hepatitis C. World J Gastroenterol. 2014;20(8): 2107-2112. CrossRef PubMed
- Ghosh S, Korza G, Maciejewski M, Setlow P. Analysis of metabolism in dormant spores of Bacillus species by 31P nuclear magnetic resonance analysis of low-molecular-weight compounds. J Bacteriol. 2015;197(5): 992-1001.PubMed
- Zhang R, Wang L, Wu F, Song B. Phosphorus speciation in the sediment profile of Lake Erhai, southwestern China: fractionation and 31P NMR. J Environ Sci (China). 2013;25(6): 1124-1130.CrossRef PubMed
- Zhang W, Jin X, Ding Y, Zhu X, Rong N, Li J, Shan B. Composition of phosphorus in wetland soils determined by SMT and solution 31P-NMR analyses. Environ Sci Pollut Res Int. 2016;23(9): 9046-9053.CrossRef PubMed
- Mani R, Waring AJ, Lehrer RI, Hong M. Membrane-disruptive abilities of beta-hairpin antimicrobial peptides correlate with conformation and activity: a 31P and 1H NMR study. Biochim Biophys Acta. 2005;1716(1): 11-18.PubMed
- Bertelsen K, Dorosz J, Hansen SK, Nielsen NC, Vosegaard T. Mechanisms of peptide-induced pore formation in lipid bilayers investigated by oriented 31P solid-state NMR spectroscopy. PLoS One. 2012;7(10): e47745.CrossRef PubMed
- Louie EA, Chirakul P, Raghunathan V, Sigurdsson ST, Drobny GP. Using solid-state 31P{19F} REDOR NMR to measure distances between a trifluoromethyl group and a phosphodiester in nucleic acids. J Magn Reson. 2006;178(1): 11-24.CrossRef PubMed
- Hallock KJ, Hamilton JA. Ex vivo identification of atherosclerotic plaque calcification by a 31P solid-state magnetic resonance imaging technique. Magn Reson Med. 2006;56(6): 1380-1383.PubMed
- Nakazawa Y, Suzuki Y, Williamson MP, Saitô H, Asakura T. The interaction of amyloid Abeta(1-40) with lipid bilayers and ganglioside as studied by 31P solid-state NMR. Chem Phys Lipids. 2009;158(1): 54-60.PubMed
- Wu Y, Reese TG, Cao H, et al. Bone mineral imaged in vivo by 31P solid state MRI of human wrists. J Magn Reson Imaging. 2011;34(3): 623-633.CrossRef PubMed
- Hobo F, Takahashi M, Saito Y, Sato N, Takao T, Koshiba S, Maeda H. 33S nuclear magnetic resonance spectroscopy of biological samples obtained with a laboratory model 33S cryogenic probe. Rev Sci Instrum. 2010;81(5): 054302.PubMed
- Ward RL, Happe JA. 35Cl NMR studies of zinc adenosine diphosphate complexes. Biochem Biophys Res Commun. 1967;28(5): 785-790.PubMed
- Cottam GL, Ward RL. 35Cl-NMR studies on zinc-pyruvate kinase complexes. Arch Biochem Biophys. 1969;132(1): 308-314.CrossRef PubMed
- Ward RL, Fritz KJ. 35Cl-NMR studies of Co2+ carbonic anhydrases. Biochem Biophys Res Commun. 1970;39(4): 707-711.CrossRef PubMed
- Norne JE, Chiancone E, Forsén S, Antonini E, Wyman J. 35Cl NMR study of the release of chloride on oxygen binding to human hemoglobin. FEBS Lett. 1978;94(2): 410-412.PubMed
- Lundberg P, Vogel H, Drakenberg T, Forsén S, Amiconi G, Forlani L, Chiancone E. A 35Cl-NMR study of the singular anion-binding properties of dromedary hemoglobin. Biochim Biophys Acta. 1989;999(1): 12-18.CrossRef PubMed
- Falke JJ, Chan SI, Steiner M, Oesterhelt D, Towner P, Lanyi JK. Halide binding by the purified halorhodopsin chromoprotein. II. New chloride-binding sites revealed by 35Cl NMR. J Biol Chem. 1984;259(4): 2185-2189.PubMed
- Koulov AV, Mahoney JM, Smith BD. Facilitated transport of sodium or potassium chloride across vesicle membranes using a ditopic salt-binding macrobicycle. Org Biomol Chem. 2003;1(1): 27-29.CrossRef PubMed
- Sidorov V, Kotch FW, Kuebler JL, Lam YF, Davis JT. Chloride transport across lipid bilayers and transmembrane potential induction by an oligophenoxyacetamide. J Am Chem Soc. 2003;125(10): 2840-2841.CrossRef PubMed
- Shachar-Hill Y, Shulman RG. Co2+ as a shift reagent for 35Cl NMR of chloride with vesicles and cells. Biochemistry. 1992;31(27): 6272-6278.PubMed
- Brauer M, Spread CY, Reithmeier RA, Sykes BD. 31P and 35Cl nuclear magnetic resonance measurements of anion transport in human erythrocytes. J Biol Chem. 1985;260(21): 11643-11650.PubMed
- Falke JJ, Kanes KJ, Chan SI. The kinetic equation for the chloride transport cycle of band 3. A 35Cl and 37Cl NMR study. J Biol Chem. 1985;260(17): 9545-9551.PubMed
- Price WS, Kuchel PW, Cornell BA. A 35Cl and 37Cl NMR study of chloride binding to the erythrocyte anion transport protein. Biophys Chem. 1991;40(3): 329-337.CrossRef PubMed
- Liu D, Knauf PA, Kennedy SD. Detection of Cl- binding to band 3 by double-quantum-filtered 35Cl nuclear magnetic resonance. Biophys J. 1996;70(2): 715-722.CrossRef PubMed
- Gettins P, Cunningham LW. A unique pair of zinc binding sites in the human alpha 2-macroglobulin tetramer. A 35Cl and 37Cl NMR study. Biochemistry. 1986;25(18): 5004-5010.PubMed
- Vetter IR, Hanssum H, Bäumert HG. Studies on the anion binding selectivity of sarcoplasmic reticulum membranes by 35Cl-NMR. Biochim Biophys Acta. 1991;1067(1): 9-16.CrossRef PubMed
- Nagel AM, Lehmann-Horn F, Weber MA, et al. In vivo 35Cl MR imaging in humans: a feasibility study. Radiology. 2014;271(2): 585-595.PubMed
- Ogino T, den Hollander JA, Shulman RG. 39K, 23Na, and 31P NMR studies of ion transport in Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1983;80(17): 5185-5189.CrossRef PubMed
- Ogino T, Shulman GI, Avison MJ, Gullans SR, den Hollander JA, Shulman RG. 23Na and 39K NMR studies of ion transport in human erythrocytes. Proc Natl Acad Sci U S A. 1985;82(4): 1099-1103. CrossRef PubMed
- Radford NB, Babcock EE, Richman A, Szczepaniak L, Malloy CR, Sherry AD. 39K NMR measurement of intracellular potassium during ischemia in the perfused guinea pig heart. Magn Reson Med. 1998;40(4): 544-550.PubMed
- Adam WR, Koretsky AP, Weiner MW. Measurement of tissue potassium in vivo using 39K nuclear magnetic resonance. Biophys J. 1987;51(2): 265-271.CrossRef PubMed
- Murakami M, Suzuki E, Miyamoto S, Seo Y, Watari H. Direct measurement of K movement by 39K NMR in perfused rat mandibular salivary gland stimulated with acetylcholine. Pflugers Arch. 1989;414(4): 385-392.PubMed
- Rashid SA, Adam WR, Craik DJ, Shehan BP, Wellard RM. Factors affecting 39K NMR detectability in rat tissue. Magn Reson Med. 1991;17(1): 213-224.PubMed
- Wellard RM, Shehan BP, Adam WR, Craik DJ. NMR measurement of 39K detectability and relaxation constants in rat tissue. Magn Reson Med. 1993;29(1): 68-76.PubMed
- Umathum R, Rösler MB, Nagel AM. In vivo 39K MR imaging of human muscle and brain. Radiology. 2013;269(2): 569-576.PubMed
- Atkinson IC, Claiborne TC, Thulborn KR. Feasibility of 39-potassium MR imaging of a human brain at 9.4 Tesla. Magn Reson Med. 2014;71(5): 1819-1825.CrossRef PubMed
- Rösler MB, Nagel AM, Umathum R, Bachert P, Benkhedah N. In vivo observation of quadrupolar splitting in 39K magnetic resonance spectroscopy of human muscle tissue. NMR Biomed. 2016;29(4): 451-457.PubMed
- Andersson T, Drakenberg T, Forsén S, Wieloch T, Lindström M. Calcium binding to porcine pancreatic prophospholipase A2 studied by 43Ca NMR. FEBS Lett. 1981;123(1): 115-117.PubMed
- Forsén S, Andersson T, Drakenberg T, Thulin E, Swärd M. Observation of the 43Ca NMR signals from Ca2+ ions bound to calmodulin, parvalbumin, and troponin C. Fed Proc. 1982;41(13): 2981-2986.PubMed
- Drakenberg T, Andersson T, Forsén S, Wieloch T. Calcium ion binding to pancreatic phospholipase A2 and its zymogen: a 43Ca NMR study. Biochemistry. 1984;23(11): 2387-2392.PubMed
- Bouhoutsos-Brown E, Pletcher CH, Nelsestuen GL, Bryant RG. Prothrombin fragment 1-membrane interactions: a calcium-43 NMR study. J Inorg Biochem. 1984;21(4): 337-343.CrossRef PubMed
- Svärd M, Drakenberg T, Andersson T, Fernlund P. Calcium binding to bone gamma-carboxyglutamic acid protein from calf studied by 43Ca NMR. Eur J Biochem. 1986;158(2): 373-378.PubMed
- Aramini JM, Drakenberg T, Hiraoki T, Ke Y, Nitta K, Vogel HJ. Calcium-43 NMR studies of calcium-binding lysozymes and alpha-lactalbumins. Biochemistry. 1992;31(29): 6761-6768.CrossRef PubMed
- Braunlin WH, Drakenberg T, Nordenskiöld L. A 43Ca-NMR study of Ca(II)-DNA interactions. Biopolymers. 1987;26(7): 1047-1062. PubMed
- Braunlin WH, Nordenskiöld L, Drakenberg T. The interaction of calcium (II) with DNA probed by 43Ca-NMR is not influenced by terminal phosphate groups at ends and nicks. Biopolymers. 1989;28(7): 1339-1342.PubMed
- Braunlin WH, Drakenberg T, Nordenskiöld L. Ca2+ binding environments on natural and synthetic polymeric DNA’s. J Biomol Struct Dyn. 1992;10(2): 333-343.CrossRef PubMed
- Wong A, Laurencin D, Wu G, Dupree R, Smith ME. An ab initio quantum chemical investigation of 43Ca NMR interaction parameters for the Ca2+ sites in organic complexes and in metalloproteins. J Phys Chem A. 2008;112(40): 9807-9813.PubMed
- Laurencin D, Gervais C, Wong A, et al. Implementation of high resolution 43Ca solid state NMR spectroscopy: toward the elucidation of calcium sites in biological materials. J Am Chem Soc. 2009;131(37): 13430-13440.PubMed
- Xu J, Zhu P, Gan Z, et al. Natural-abundance 43Ca solid-state NMR spectroscopy of bone. J Am Chem Soc. 2010;132(33): 11504-11509. PubMed
- Willsky GR, Dosch SF. Vanadium metabolism in wild type and respiratory-deficient strains of S. cerevisiae. Yeast. 1986;2(2): 77-85. PubMed
- Rehder D, Holst H, Quaas R, Hinrichs W, Hahn U, Saenger W. Binding of vanadate (V) to ribonuclease-T1 and inosine, investigated by 51V NMR spectroscopy. J Inorg Biochem. 1989;37(2): 141-150.PubMed
- Liu S, Gresser MJ, Tracey AS. 1H and 51V NMR studies of the interaction of vanadate and 2-vanadio-3-phosphoglycerate with phosphoglycerate mutase. Biochemistry. 1992;31(10): 2677-2685. PubMed
- Hochman Y, Carmeli S, Carmeli C. Vanadate, a transition state inhibitor of chloroplast CF1-ATPase. J Biol Chem. 1993;268(17): 12373-12379.PubMed
- Saponja JA, Vogel HJ. Metal-ion binding properties of the transferrins: a vanadium-51 NMR study. J Inorg Biochem. 1996;62(4): 253-270.CrossRef PubMed
- Rehder D, Casný M, Grosse R. A vanadium-51 NMR study of the binding of vanadate and peroxovanadate to proteins. Magn Reson Chem. 2004;42(9): 745-749.CrossRef PubMed
- Aureliano M, Mdeira VM. Interactions of vanadate oligomers with sarcoplasmic reticulum Ca2+-ATPase. Biochim Biophys Acta. 1994;1221(3): 259-271.CrossRef PubMed
- Zhang B, Ruan L, Chen B, Lu J, Wang K. Binding of vanadate to human erythrocyte ghosts and subsequent events. Biometals. 1997;10(4): 291-298.PubMed
- Pooransingh-Margolis N, Renirie R, Hasan Z, Wever R, Vega AJ, Polenova T. 51V solid-state magic angle spinning NMR spectroscopy of vanadium chloroperoxidase. J Am Chem Soc. 2006;128(15): 5190-5208.CrossRef PubMed
- Fenn A, Wächtler M, Gutmann T, et al. Correlations between 51V solid-state NMR parameters and chemical structure of vanadium (V) complexes as models for related metalloproteins and catalysts. Solid State Nucl Magn Reson. 2009;36(4): 192-201.CrossRef PubMed
- Fenn A, Wächtler M, Breitzke H, Buchholz A, Lippold I, Plass W, Buntkowsky G. A simple method for analyzing 51V solid-state NMR spectra of complex systems. Solid State Nucl Magn Reson. 2011;40(2): 60-65.CrossRef PubMed
- Shinar H, Navon G. The determination of intracellular water space by NMR. FEBS Lett. 1985;193(1): 75-78.CrossRef PubMed
- Kushnir T, Kaplan O, Askenasy N, Navon G. 23Na, 59Co and 2H NMR studies of experimental acute pancreatitis. NMR Biomed. 1991;4(4): 182-186.PubMed
- Askenasy N, Navon G. Measurements of intracellular volumes by 59Co and 2H/1H NMR and their physiological applications. NMR Biomed. 2005;18(2): 104-110.PubMed
- Webb AG, Wong M, Niesman M, Kolbeck KJ, Wilmes LJ, Magin RL, Suslick KS. In vivo NMR thermometry with liposomes containing 59Co complexes. Int J Hyperthermia. 1995;11(6): 821-827.PubMed
- Xu Q, Jampani SR, Deng H, Braunlin WH. Chiral recognition of deoxyoligonucleotides by delta- and lambda-tris(ethylenediamine)cobalt(III). Biochemistry. 1995;34(43): 14059-14065.CrossRef PubMed
- Grant CV, Frydman V, Harwood JS, Frydman L. 59Co solid-state NMR as a new probe for elucidating metal binding in polynucleotides. J Am Chem Soc. 2002;124(16): 4458-4462.CrossRef PubMed
- Lipton AS, Heck RW, de Jong WA, et al. Low temperature 65Cu NMR spectroscopy of the Cu+ site in azurin. J Am Chem Soc. 2009;131(39): 13992-13999.PubMed
- Lipton AS, Buchko GW, Sears JA, Kennedy MA, Ellis PD. 67Zn solid-state NMR spectroscopy of the minimal DNA binding domain of human nucleotide excision repair protein XPA. J Am Chem Soc. 2001;123(5): 992-993.CrossRef PubMed
- Lipton AS, Heck RW, Staeheli GR, Valiev M, De Jong WA, Ellis PD. A QM/MM approach to interpreting 67Zn solid-state NMR data in zinc proteins. J Am Chem Soc. 2008;130(19): 6224-6230.PubMed
- Lipton AS, Heck RW, Hernick M, Fierke CA, Ellis PD. Residue ionization in LpxC directly observed by 67Zn NMR spectroscopy. J Am Chem Soc. 2008;130(38): 12671-12679.PubMed
- Luthra NP, Costello RC, Odom JD, Dunlap RB. Demonstration of the feasibility of observing nuclear magnetic resonance signals of 77Se covalently attached to proteins. J Biol Chem. 1982;257(3): 1142-1144.PubMed
- Gettins P, Wardlaw SA. NMR relaxation properties of 77Se-labeled proteins. J Biol Chem. 1991;266(6): 3422-3426.PubMed
- Gettins P, Crews BC. 77Se NMR characterization of 77Se-labeled ovine erythrocyte glutathione peroxidase. J Biol Chem. 1991;266(8): 4804-4809.PubMed
- Mobli M, Morgenstern D, King GF, Alewood PF, Muttenthaler M. Site-specific pKa determination of selenocysteine residues in selenovasopressin by using 77Se NMR spectroscopy. Angew Chem Int Ed Engl. 2011;50(50): 11952-11955.CrossRef PubMed
- Garnett MW, Halstead TK, Hoare DG. Study of the interaction of 4-bromomercuriocinnamic acid with alpha-chymotrypsin by 79Br and 81Br pulsed nuclear-magnetic resonance. Eur J Biochem. 1976;66(1): 85-93.CrossRef PubMed
- Pavlovskaya GE, Cleveland ZI, Stupic KF, Basaraba RJ, Meersmann T. Hyperpolarized krypton-83 as a contrast agent for magnetic resonance imaging. Proc Natl Acad Sci U S A. 2005;102(51): 18275-18279.CrossRef PubMed
- Lilburn DM, Lesbats C, Six JS, et al. Hyperpolarized 83Kr magnetic resonance imaging of alveolar degradation in a rat model of emphysema. J R Soc Interface. 2015;12(107): 20150192.CrossRef PubMed
- Cleveland ZI, Pavlovskaya GE, Elkins ND, Stupic KF, Repine JE, Meersmann T. Hyperpolarized 83Kr MRI of lungs. J Magn Reson. 2008;195(2): 232-237.CrossRef PubMed
- Cleveland ZI, Pavlovskaya GE, Stupic KF, Wooten JB, Repine JE, Meersmann T. Detection of tobacco smoke deposition by hyperpolarized krypton-83 MRI. Magn Reson Imaging. 2008;26(2): 270-278.CrossRef PubMed
- Helpern JA, Welch KM, Halvorson HR. Rubidium transport in human erythrocyte suspensions monitored by 87Rb NMR with aqueous chemical shift reagents. NMR Biomed. 1989;2(2): 47-54.PubMed
- Kupriyanov VV, Gruwel ML. Rubidium-87 magnetic resonance spectroscopy and imaging for analysis of mammalian K+ transport. NMR Biomed. 2005;18(2): 111-124.CrossRef PubMed
- Kupriyanov VV, Shen J, Xiang B, Kuzio B, Sun J, Deslauriers R. Three-dimensional 87Rb imaging of isolated pig hearts: effects of regional ischemia. Magn Reson Med. 1998;40(2): 175-179.PubMed
- Gruwel ML, Kuzio B, Deslauriers R, Kupriyanov VV. Measurements of mitochondrial K+ fluxes in whole rat hearts using 87Rb-NMR. Am J Physiol. 1999;276(1): C193-C200.PubMed
- Jilkina O, Xiang B, Kuzio B, Rendell J, Kupriyanov VV. Potassium transport in Langendorff-perfused mouse hearts assessed by 87Rb NMR spectroscopy. Magn Reson Med. 2005;53(5): 1172-1176.PubMed
- Kupriyanov VV, Xiang B, Sun J, Jilkina O, Kuzio B. Imaging of ischemia and infarction in blood-perfused pig hearts using 87Rb MRI. Magn Reson Med. 2003;49(1): 99-107.PubMed
- Jilkina O, Kuzio B, Rendell J, Xiang B, Kupriyanov VV. K+ transport and energetics in Kir6.2(-/-) mouse hearts assessed by 87Rb and 31P magnetic resonance and optical spectroscopy. J Mol Cell Cardiol. 2006;41(5): 893-901.CrossRef PubMed
- Blackburn GM, Mann BE, Taylor BF, Worrall AF. A nuclear-magnetic-resonance study of the binding of novel N-hydroxybenzenesulphonamide carbonic anhydrase inhibitors to native and cadmium-111-substituted carbonic anhydrase. Eur J Biochem. 1985;153(3): 553-558.CrossRef PubMed
- Mekmouche Y, Coppel Y, Hochgräfe K, Guilloreau L, Talmard C, Mazarguil H, Faller P. Characterization of the ZnII binding to the peptide amyloid-beta1-16 linked to Alzheimer’s disease. Chembiochem. 2005;6(9): 1663-1671.PubMed
- Li H, Otvos JD. 111Cd NMR studies of the domain specificity of Ag+ and Cu+ binding to metallothionein. Biochemistry. 1996;35(44): 13929-13936.CrossRef PubMed
- Daniels MJ, Turner-Cavet JS, Selkirk R, Sun H, Parkinson JA, Sadler PJ, Robinson NJ. Coordination of Zn2+ (and Cd2+) by prokaryotic metallothionein. Involvement of his-imidazole. J Biol Chem. 1998;273(36): 22957-22961.CrossRef PubMed
- Leszczyszyn OI, Schmid R, Blindauer CA. Toward a property/function relationship for metallothioneins: histidine coordination and unusual cluster composition in a zinc-metallothionein from plants. Proteins. 2007;68(4): 922-935.CrossRef PubMed
- Vasák M. Application of 113Cd NMR to metallothioneins. Biodegradation. 1998;9(6): 501-512.PubMed
- Armitage IM, Drakenberg T, Reilly B. Use of 113Cd NMR to probe the native metal binding sites in metalloproteins: an overview. Met Ions Life Sci. 2013;11: 117-144.CrossRef PubMed
- Armitage IM, Otvos JD, Briggs RW, Boulanger Y. Structure elucidation of the metal-binding sites in metallothionein by 113Cd NMR. Fed Proc. 1982;41(13): 2974-2980.PubMed
- Wagner G, Frey MH, Neuhaus D, et al. Spatial structure of rabbit liver metallothionein-2 in solution by NMR. Experientia Suppl. 1987;52: 149-157.CrossRef PubMed
- Cismowski MJ, Narula SS, Armitage IM, Chernaik ML, Huang PC. Mutation of invariant cysteines of mammalian metallothionein alters metal binding capacity, cadmium resistance, and 113Cd NMR spectrum. J Biol Chem. 1991;266(36): 24390-24397.PubMed
- Narula SS, Brouwer M, Hua Y, Armitage IM. Three-dimensional solution structure of Callinectes sapidus metallothionein-1 determined by homonuclear and heteronuclear magnetic resonance spectroscopy. Biochemistry. 1995;34(2): 620-631.CrossRef PubMed
- Wang H, Li H, Cai B, Huang ZX, Sun H. The effect of nitric oxide on metal release from metallothionein-3: gradual unfolding of the protein. J Biol Inorg Chem. 2008;13(3): 411-419.CrossRef PubMed
- Karotki AV, Vasák M. Reaction of human metallothionein-3 with cisplatin and transplatin. J Biol Inorg Chem. 2009;14(7): 1129-1138. CrossRef PubMed
- Jonsson NB, Tibell LA, Evelhoch JL, Bell SJ, Sudmeier JL. Cadmium-113 NMR of carbonic anhydrases: effect of pH, bicarbonate, and cyanide. Proc Natl Acad Sci U S A. 1980;77(6): 3269-3272.CrossRef PubMed
- Engeseth HR, McMillin DR, Otvos JD. Comparative Cd-113 nuclear magnetic resonance studies of Cd(II)-substituted blue copper proteins. J Biol Chem. 1984;259(8): 4822-4826.PubMed
- Kofod P, Bauer R, Danielsen E, Larsen E, Bjerrum MJ. 113Cd-NMR investigation of a cadmium-substituted copper, zinc-containing superoxide dismutase from yeast. Eur J Biochem. 1991;198(3): 607-611.CrossRef PubMed
- Knegtel RM, Boelens R, Ganadu ML, George AV, van der Saag PT, Kaptein R. Heteronuclear 113Cd-1H NMR study of metal coordination in the human retinoic acid receptor-beta DNA binding domain. Biochem Biophys Res Commun. 1993;192(2): 492-498.PubMed
- Omburo GA, Mullins LS, Raushel FM. Structural characterization of the divalent cation sites of bacterial phosphotriesterase by 113Cd NMR spectroscopy. Biochemistry. 1993;32(35): 9148-9155.PubMed
- Aramini JM, Hiraoki T, Ke Y, Nitta K, Vogel HJ. Cadmium-113 NMR studies of bovine and human alpha-lactalbumin and equine lysozyme. J Biochem. 1995;117(3): 623-628.PubMed
- Kanaori K, Ohta D, Nosaka AY. Effect of excess cadmium ion on the metal binding site of cabbage histidinol dehydrogenase studied by 113Cd-NMR spectroscopy. FEBS Lett. 1997;412(2): 301-304.PubMed
- Kellenbach E, Maler BA, Yamamoto KR, Boelens R, Kaptein R. Identification of the metal coordinating residues in the DNA binding domain of the glucocorticoid receptor by 113Cd-1H heteronuclear NMR spectroscopy. FEBS Lett. 1991;291(2): 367-370.PubMed
- Ueno D, Ma JF, Iwashita T, Zhao FJ, McGrath SP. Identification of the form of Cd in the leaves of a superior Cd-accumulating ecotype of Thlaspi caerulescens using 113Cd-NMR. Planta. 2005;221(6): 928-936.CrossRef PubMed
- Ueno D, Iwashita T, Zhao FJ, Ma JF. Characterization of Cd translocation and identification of the Cd form in xylem sap of the Cd-hyperaccumulator Arabidopsis halleri. Plant Cell Physiol. 2008;49(4): 540-548.CrossRef PubMed
- Grassi M, Mingazzini M. 113Cd-NMR and fluorescence studies of the interactions between Cd(II) and extracellular organic matter released by Selenastrum capricornutum. Environ Sci Technol. 2001;35(21): 4271-4276.PubMed
- Lenoble V, Garnier C, Masion A, Ziarelli F, Garnier JM. Combination of 13C/ 113Cd NMR, potentiometry, and voltammetry in characterizing the interactions between Cd and two models of the main components of soil organic matter. Anal Bioanal Chem. 2008;390(2): 749-757.CrossRef PubMed
- Rahman M, Patching SG, Ismat F, et al. Probing metal ion substrate-binding to the E. coli ZitB exporter in native membranes by solid state NMR. Mol Membr Biol. 2008;25(8): 683-690.PubMed
- Sakurada J, Takahashi S, Shimizu T, Hatano M, Nakamura S, Hosoya T. Proton and iodine-127 nuclear magnetic resonance studies on the binding of iodide by lactoperoxidase. Biochemistry. 1987;26(20): 6478-6483.CrossRef PubMed
- Fukuyama K1, Sato K, Itakura H, Takahashi S, Hosoya T. Binding of iodide to Arthromyces ramosus peroxidase investigated with X-ray crystallographic analysis, 1H and 127I NMR spectroscopy, and steady-state kinetics. J Biol Chem. 1997;272(9): 5752-5756. CrossRef PubMed
- Oros AM, Shah NJ. Hyperpolarized xenon in NMR and MRI. Phys Med Biol. 2004;49(20): R105-R153.CrossRef PubMed
- Aaron JA, Chambers JM, Jude KM, Di Costanzo L, Dmochowski IJ, Christianson DW. Structure of a 129Xe-cryptophane biosensor complexed with human carbonic anhydrase II. J Am Chem Soc. 2008;130(22): 6942-6943.PubMed
- Taratula O, Bai Y, D’Antonio EL, Dmochowski IJ. Enantiopure cryptophane-129Xe nuclear magnetic resonance biosensors targeting carbonic anhydrase. Supramol Chem. 2015;27(1-2): 65-71.CrossRef PubMed
- Boutin C, Stopin A, Lenda F, et al. Cell uptake of a biosensor detected by hyperpolarized 129Xe NMR: the transferrin case. Bioorg Med Chem. 2011;19(13): 4135-4143.CrossRef PubMed
- Seward GK, Bai Y, Khan NS, Dmochowski IJ. Cell-compatible, integrin-targeted cryptophane-129Xe NMR biosensors. Chem Sci. 2011;2(6): 1103-1110.PubMed
- Delacour L, Kotera N, Traoré T, et al. “Clickable” hydrosoluble PEGylated cryptophane as a universal platform for 129Xe magnetic resonance imaging biosensors. Chemistry. 2013;19(19): 6089-6093.PubMed
- Palaniappan KK, Ramirez RM, Bajaj VS, Wemmer DE, Pines A, Francis MB. Molecular imaging of cancer cells using a bacteriophage-based 129Xe NMR biosensor. Angew Chem Int Ed Engl. 2013;52(18): 4849-4853.PubMed
- Rose HM, Witte C, Rossella F, Klippel S, Freund C, Schröder L. Development of an antibody-based, modular biosensor for 129Xe NMR molecular imaging of cells at nanomolar concentrations. Proc Natl Acad Sci U S A. 2014;111(32): 11697-11702.CrossRef PubMed
- Bai Y, Wang Y, Goulian M, Driks A, Dmochowski IJ. Bacterial spore detection and analysis using hyperpolarized 129Xe chemical exchange saturation transfer (Hyper-CEST) NMR. Chem Sci. 2014;5(8): 3197-3203.CrossRef PubMed
- Tassali N, Kotera N, Boutin C, et al. Smart detection of toxic metal ions, Pb2+ and Cd2+, using a 129Xe NMR-based sensor. Anal Chem. 2014;86(3): 1783-1788.PubMed
- Boutin C, Léonce E, Brotin T, Jerschow A, Berthault P. Ultrafast Z-spectroscopy for 129Xe NMR-based sensors. J Phys Chem Lett. 2013;4(23): 4172-4176.PubMed
- Jiménez-Martínez R, Kennedy DJ, Rosenbluh M, et al. Optical hyperpolarization and NMR detection of 129Xe on a microfluidic chip. Nat Commun. 2014;5: 3908.PubMed
- Mugler JP 3rd, Altes TA. Hyperpolarized 129Xe MRI of the human lung. J Magn Reson Imaging. 2013;37(2): 313-331.PubMed
- Kirby M, Svenningsen S, Owrangi A, et al. Hyperpolarized 3He and 129Xe MR imaging in healthy volunteers and patients with chronic obstructive pulmonary disease. Radiology. 2012;265(2): 600-610.PubMed
- Chang YV, Quirk JD, Ruset IC, Atkinson JJ, Hersman FW, Woods JC. Quantification of human lung structure and physiology using hyperpolarized 129Xe. Magn Reson Med. 2014;71(1): 339-344. PubMed
- Qing K, Ruppert K, Jiang Y, et al. Regional mapping of gas uptake by blood and tissue in the human lung using hyperpolarized xenon-129 MRI. J Magn Reson Imaging. 2014;39(2): 346-359.CrossRef PubMed
- Zhou X, Sun Y, Mazzanti M, Henninger N, Mansour J, Fisher M, Albert M. MRI of stroke using hyperpolarized 129Xe. NMR Biomed. 2011;24(2): 170-175.CrossRef PubMed
- Branca RT, He T, Zhang L, Floyd CS, Freeman M, White C, Burant A. Detection of brown adipose tissue and thermogenic activity in mice by hyperpolarized xenon MRI. Proc Natl Acad Sci U S A. 2014;111(50): 18001-18006.CrossRef PubMed
- Li H, Zhang Z, Zhong J, et al. Oxygen-dependent hyperpolarized 129Xe brain MR. NMR Biomed. 2016;29(3): 220-225.PubMed
- Goodman J, Neil JJ, Ackerman JJ. Biomedical applications of 133Cs NMR. NMR Biomed. 2005;18(2): 125-134.PubMed
- Shehan BP, Wellard RM, Adam WR, Craik DJ. The use of dietary loading of 133Cs as a potassium substitute in NMR studies of tissues. Magn Reson Med. 1993;30(5): 573-582.PubMed
- Davis DG, Murphy E, London RE. Uptake of cesium ions by human erythrocytes and perfused rat heart: a cesium-133 NMR study. Biochemistry. 1988;27(10): 3547-3551.CrossRef PubMed
- Bramham J, Riddell FG. Cesium uptake studies on human erythrocytes. J Inorg Biochem. 1994;53(3): 169-176.CrossRef PubMed
- Pfeffer PE, Rolin DB, Brauer D, Tu SI, Kumosinski TF. In vivo 133Cs-NMR a probe for studying subcellular compartmentation and ion uptake in maize root tissue. Biochim Biophys Acta. 1990;1054(2): 169-175.CrossRef PubMed
- Li Y, Neil J, Ackerman JJ. On the use of 133Cs as an NMR active probe of intracellular space in vivo. NMR Biomed. 1995;8(5): 183-189.PubMed
- Schornack PA, Song SK, Ling CS, Hotchkiss R, Ackerman JJ. Quantification of ion transport in perfused rat heart: 133Cs+ as an NMR active K+ analog. Am J Physiol. 1997;272(5): C1618-C1634.PubMed
- Schornack PA, Song SK, Hotchkiss R, Ackerman JJ. Inhibition of ion transport in septic rat heart: 133Cs+ as an NMR active K+ analog. Am J Physiol. 1997;272(5): C1635-C1641.PubMed
- Gruwel ML, Williams JP. Short-term regulation of endothelial Na+-K+-pump activity by cGMP: a 133Cs magnetic resonance study. Mol Membr Biol. 1998;15(4): 189-192.CrossRef PubMed
- Wellard RM, Adam WR. Functional hepatocyte cation compartmentation demonstrated with 133Cs NMR. Magn Reson Med. 2002;48(5): 810-818.PubMed
- Le Lay P, Isaure MP, Sarry JE, et al. Metabolomic, proteomic and biophysical analyses of Arabidopsis thaliana cells exposed to a caesium stress. Influence of potassium supply. Biochimie. 2006;88(11): 1533-1547.CrossRef PubMed
- Pham BT, Jain N, Kuchel PW, Chapman BE, Bickley SA, Jones SK, Hawkett BS. The interaction of sterically stabilized magnetic nanoparticles with fresh human red blood cells. Int J Nanomedicine. 2015;10: 6645-6655.PubMed
- Pendyala L, Krishnan BS, Walsh JR, Arakali AV, Cowens JW, Creaven PJ. Studies on the human metabolism of iproplatin. Cancer Chemother Pharmacol. 1989;25(1): 10-14.CrossRef PubMed
- Becker M, Port RE, Zabel HJ, Zeller WJ, Bachert P. Monitoring local disposition kinetics of carboplatin in vivo after subcutaneous injection in rats by means of 195Pt NMR. J Magn Reson. 1998;133(1): 115-122.PubMed
- Delnomdedieu M, Boudou A, Georgescauld D, Dufourc EJ. Specific interactions of mercury chloride with membranes and other ligands as revealed by mercury-NMR. Chem Biol Interact. 1992;81(3): 243-269.CrossRef PubMed
- Utschig LM, Bryson JW, O’Halloran TV. Mercury-199 NMR of the metal receptor site in MerR and its protein-DNA complex. Science. 1995;268(5209): 380-385.CrossRef PubMed
- Steele RA, Opella SJ. Structures of the reduced and mercury-bound forms of MerP, the periplasmic protein from the bacterial mercury detoxification system. Biochemistry. 1997;36(23): 6885-6895. CrossRef PubMed
- Hinton JF, Koeppe RE 2nd, Shungu D, Whaley WL, Paczkowski JA, Millett FS. Equilibrium binding constants for Tl+ with gramicidins A, B and C in a lysophosphatidylcholine environment determined by 205Tl nuclear magnetic resonance spectroscopy. Biophys J. 1986;49(2): 571-577.CrossRef PubMed
- Shungu DC, Hinton JF, Koeppe RE 2nd, Millett FS. Investigation of the interaction between thallous ions and gramicidin A in dimyristoylphosphatidylcholine vesicles: a thallium-205 NMR equilibrium study. Biochemistry. 1986;25(20): 6103-6108.CrossRef PubMed
- Markham GD. Characterization of the monovalent cation activator binding site of S-adenosylmethionine synthetase by 205Tl NMR of enzyme-bound Tl+. J Biol Chem. 1986;261(4): 1507-1509.PubMed
- Hill KA, Steiner SA, Castellino FJ. 205Tl+ as a spectroscopic probe of the monovalent cation binding sites of bovine plasma activated protein C and des-1-41-light-chain-activated protein C. J Biol Chem. 1987;262(15): 7098-7104.PubMed
- Aramini JM, Krygsman PH, Vogel HJ. Thallium-205 and carbon-13 NMR studies of human sero- and chicken ovotransferrin. Biochemistry. 1994;33(11): 3304-3311.CrossRef PubMed
- Loria JP, Nowak T. Conformational changes in yeast pyruvate kinase studied by 205Tl+ NMR. Biochemistry. 1998;37(19): 6967-6974.PubMed
- Susan-Resiga D, Nowak T. Monitoring active site alterations upon mutation of yeast pyruvate kinase using 205Tl+ NMR. J Biol Chem. 2003;278(42): 40943-40952.CrossRef PubMed
- Gill ML, Strobel SA, Loria JP. 205Tl NMR methods for the characterization of monovalent cation binding to nucleic acids. J Am Chem Soc. 2005;127(47): 16723-16732.CrossRef PubMed
- Jakobsen LO, Malmendal A, Nielsen NC, Esmann M. Cation binding in Na,K-ATPase, investigated by 205Tl solid-state NMR spectroscopy. Biochemistry. 2006;45(35): 10768-10776.PubMed
- Neupane KP, Pecoraro VL. Pb-207 NMR spectroscopy reveals that Pb(II) coordinates with glutathione (GSH) and tris cysteine zinc finger proteins in a PbS3 coordination environment. J Inorg Biochem. 2011;105(8): 1030-1034.CrossRef PubMed
Article information
Corresponding author: Simon G. Patching.
Copyright: © 2016 Patching SG. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are cited.
How to Cite: Patching SG. NMR active nuclei for biological and biomedical applications. J. Diagn. Imaging Ther. 2016;3 (1): 7-48 (https://dx.doi.org/10.17229/jdit.2016-0618-021).
Article history: Received 9 May 2016; Accepted 25 May 2016; Published online 17 June 2016.
Archive link: JDIT-2016-0618-021

You Are Here: Home »